
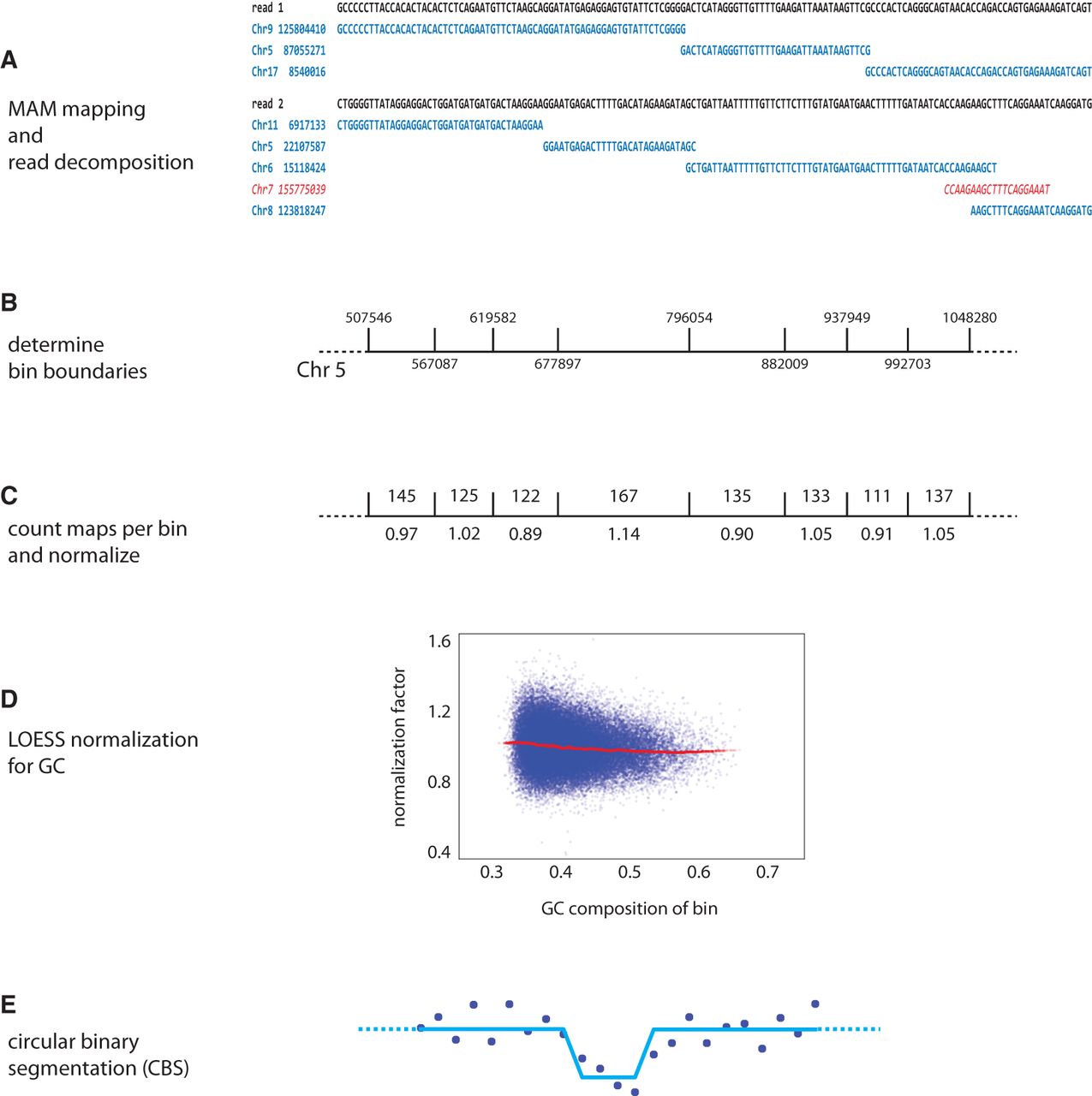
SMASH informatics pipeline. (A) The decomposition of a read pair into a set of maximal uniquely mappable fragments is shown. In contrast to the red maps, the blue maps satisfy the 20:4 rule and are considered countable maps. (B) Bin boundaries are selected such that each bin has the same number of exact matches from all 50-mers from the reference genome. A representative stretch of Chromosome 5 is displayed. (C) The numbers of 20:4 mappable fragments present in each bin are counted, with duplicate reads excluded. The number above the bin shows the count of maps, and the number below shows the normalized value. (D) LOESS normalization is used to adjust bin counts for sample-specific GC bias. (E) The data are segmented using circular binary segmentation (CBS) of the GC-normalized data.








