
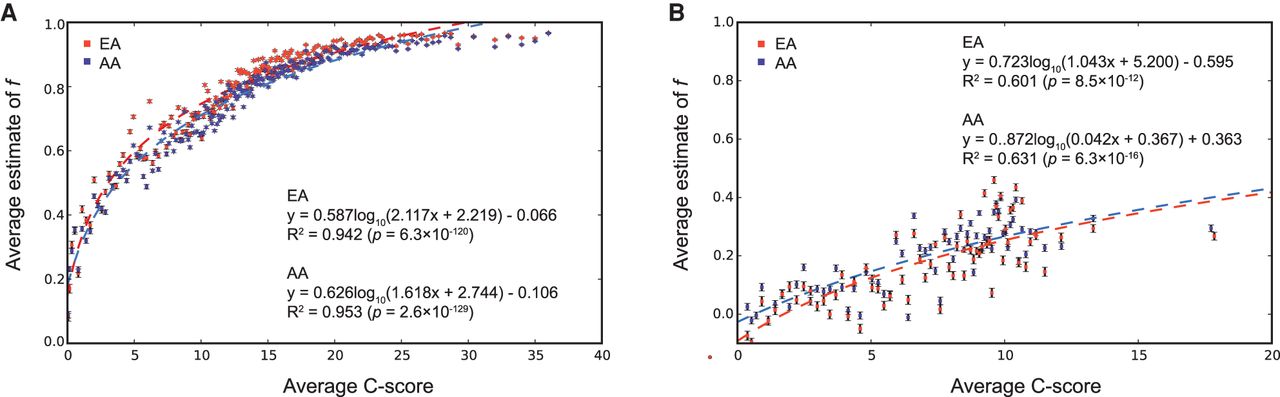
Figure 5.
Relationship between predicted pathogenicity of SNVs and estimate of f in populations. The average estimate of f on nonsynonymous (A) and synonymous (B) SNVs was computed on each bootstrapped subset of SNVs in each bin by using a quantile-binning approach, such that each bin had the same number of SNVs. Error bars, SEM scaled C-score (x-axis) and mean estimate of f (y-axis). The red (EA) and blue (AA) lines represent the best fit curves to the data.








