
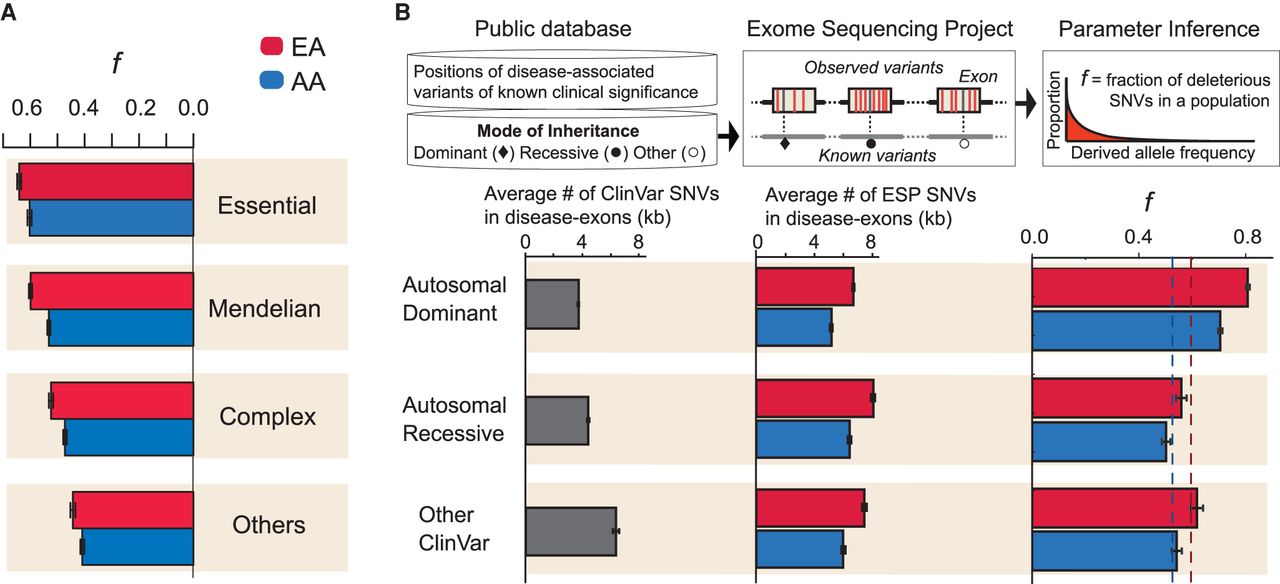
Intensity of purifying selection on nonsynonymous SNVs in disease related genes. (A) Estimates of f in genes designated as Essential, Mendelian, Complex, or Others (for details, see text). (B) Estimates of f from SNVs in exons with reported disease causing mutations as a function of mode of inheritance. The top panel provides a schematic illustration of the analysis, including intersection between exons containing ClinVar reported mutations and nonsynonymous SNVs in ESP. The plots below show the number of nonsynonymous SNVs/kb estimated from ClinVar and ESP, as well as estimates of f as a function of mode of inheritance. Dashed lines denote the average f for nonsynonymous SNVs in EAs (red) and AAs (blue).








