
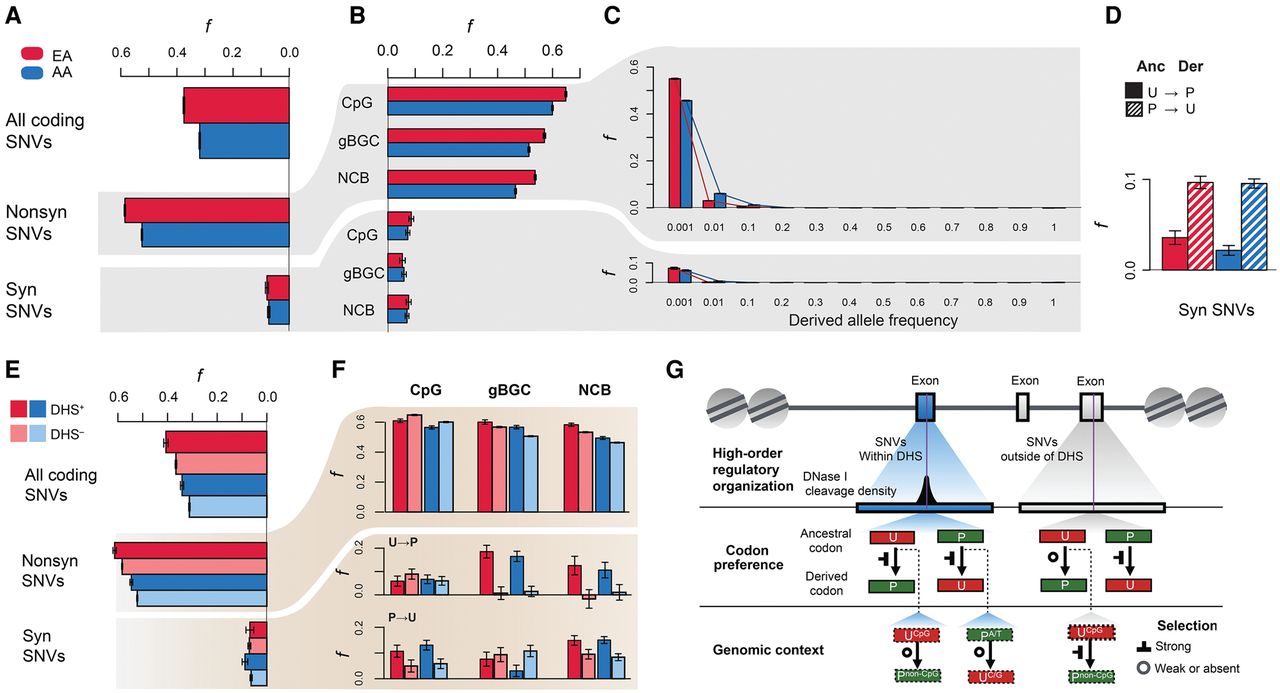
Characteristics of deleterious protein-coding SNVs. Estimates of f for (A) all protein-coding SNVs, all nonsynonymous SNVs, and all synonymous SNVs in EAs (red) and AAs (blue). (B) Estimates of f for SNVs as a function of genomic context. (C) Estimates of f as a function of derived allele frequency. (D) Effect of codon bias on estimates of f at synonymous sites. (E) Estimates of f for SNVs inside (+; darker shade) or outside (−; lighter shade) of DHSs. (F) Decomposing the effects of genomic context, codon preference, and DHSs on estimates of f. (G) Schematic summary of context-dependent patterns of deleterious synonymous SNVs (purple, vertical lines) as a function of regulatory context (first row), codon preference (second row), and genomic context (third row). Note that preferred changes (U → P) at CpG sites and unpreferred changes (P → U) of gBGC within the DHSs show less constraint, whereas preferred changes (U → P) at CpG sites outside of DHSs exhibit stronger constraint.








