
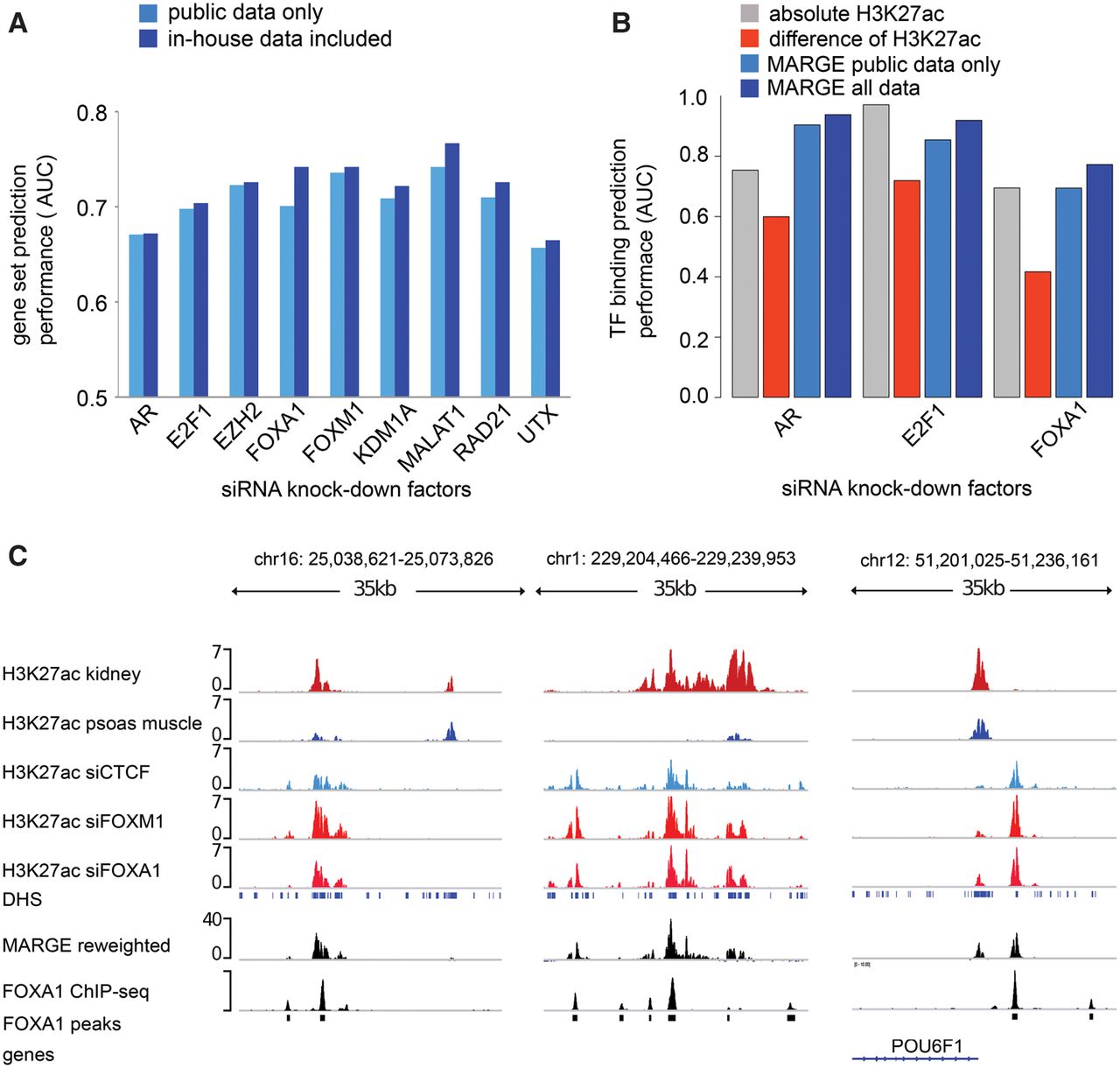
MARGE-cistrome prediction of cis-regulatory regions from knockdown gene expression and H3K27ac ChIP-seq data. (A) Down-regulated genes in LNCaP-abl prostate cancer cells on siRNA silencing of nine factors can be predicted from the compendium of H3K27ac ChIP-seq profiles. Augmentation of public data with H3K27ac ChIP-seq generated in LNCaP-abl samples improves prediction performance slightly. (B) Prediction of AR, E2F1, and FOXA1 binding sites using four methods: sample-specific H3K27ac ChIP-seq read count; difference of square root H3K27ac read counts between wild-type and knockdown samples; MARGE-cistrome based on public H3K27ac ChIP-seq data only; MARGE-cistrome based on public data augmented with H3K27ac ChIP-seq data in LNCaP-abl. (C) Example of predicted cis-regulatory loci with FOXA1 binding sites. The MARGE reweighted track is a linear combination of H3K27ac signal tracks with coefficients defined by MARGE-cistrome.








