
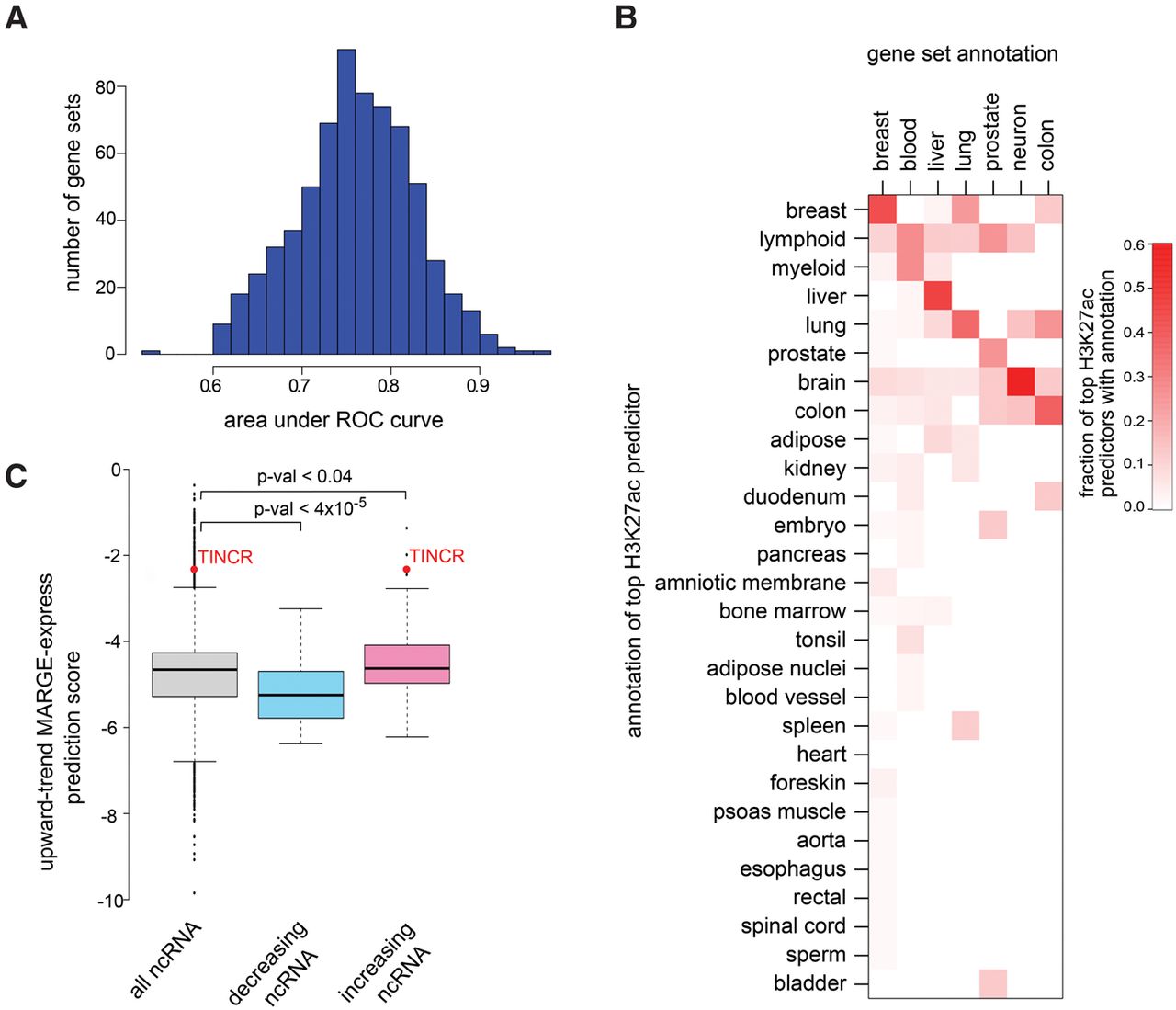
MARGE-express modeling of differential expression gene sets using H3K27ac regulatory potentials in diverse samples. (A) An analysis of 671 gene sets from MSigDB shows, using independent training and testing data, that this approach is highly predictive of most gene sets. (B) Heat map of the proportion, by tissue type, of H3K27ac samples that are most predictive of the tissue type-associated gene sets. Gene sets with descriptions that include the keyword liver, for example, are most often predicted by liver-derived H3K27ac ChIP-seq samples. In this example, the fraction of times that liver-derived H3K27ac samples are selected first in the step-wise regression analysis is represented in the liver-associated row of this heat map. (C) MARGE-express prediction of differentially expressed noncoding RNAs based on coding RNA data. A gene set based on the upward trending protein-coding genes in a time course of keratinocyte differentiation was used as input data. MARGE-express predicted scores for noncoding RefSeq genes. These scores are compared between upward and downward trending noncoding RNAs observed in a separate keratinocyte differentiation experiment. TINCR is a strongly up-regulated lncRNA in the differentiated state and is especially important to keratinocyte development.








