
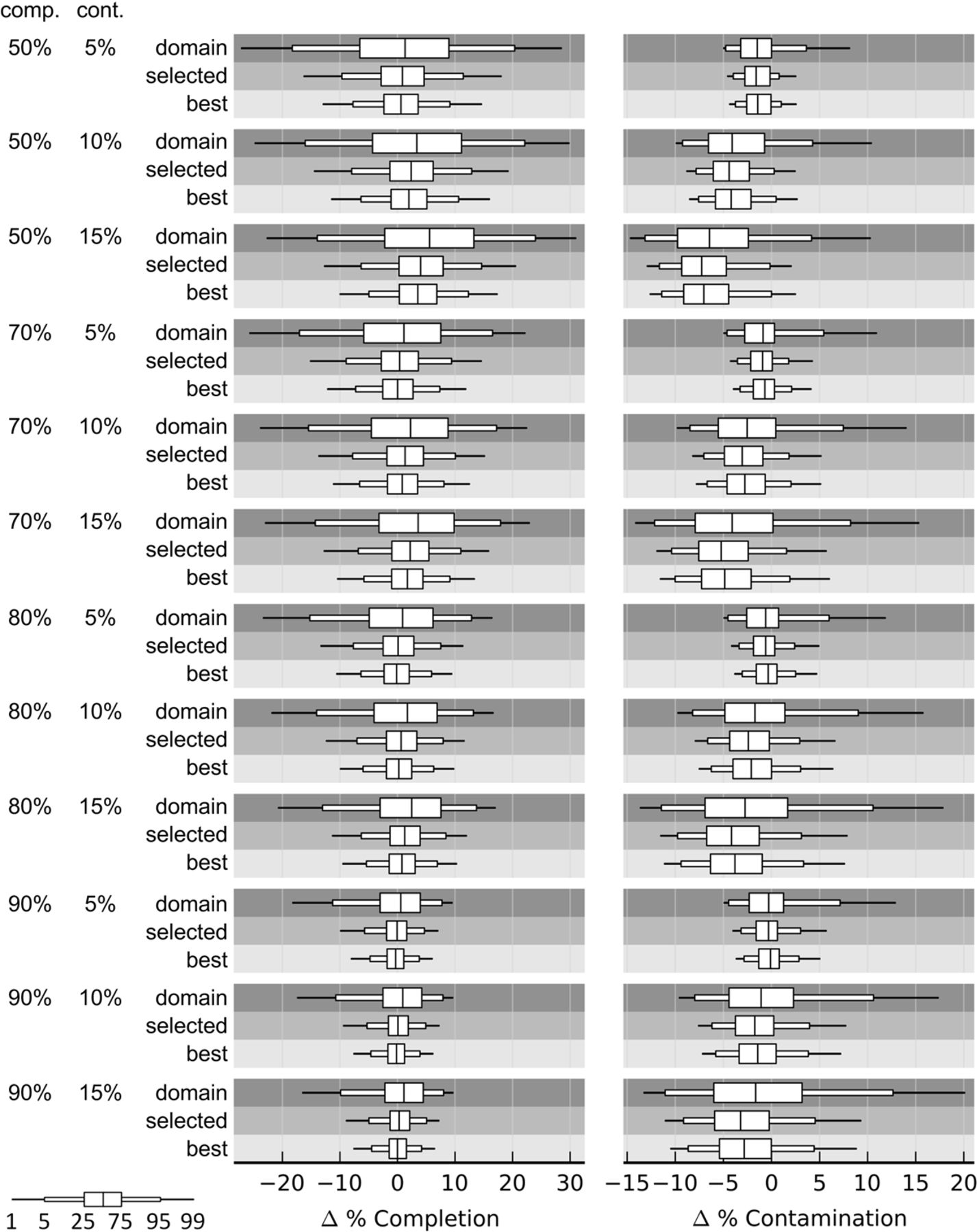
Error in completeness and contamination estimates on simulated genomes with 50%, 70%, 80%, or 90% completeness and 5%, 10%, or 15% contamination. Quality estimates were determined using (1) domain: marker sets inferred across all archaeal or bacterial genomes; (2) selected: marker sets inferred from genomes within the lineage selected by CheckM; and (3) best: marker sets inferred from genomes within the lineage producing the most accurate estimates. Marker genes were organized into collocated marker sets in all cases. Simulated genomes were generated under the random contig model from 2430 draft genomes spanning 31 classes (18 phyla) with each draft genome being used to generate 20 simulated genomes.








