
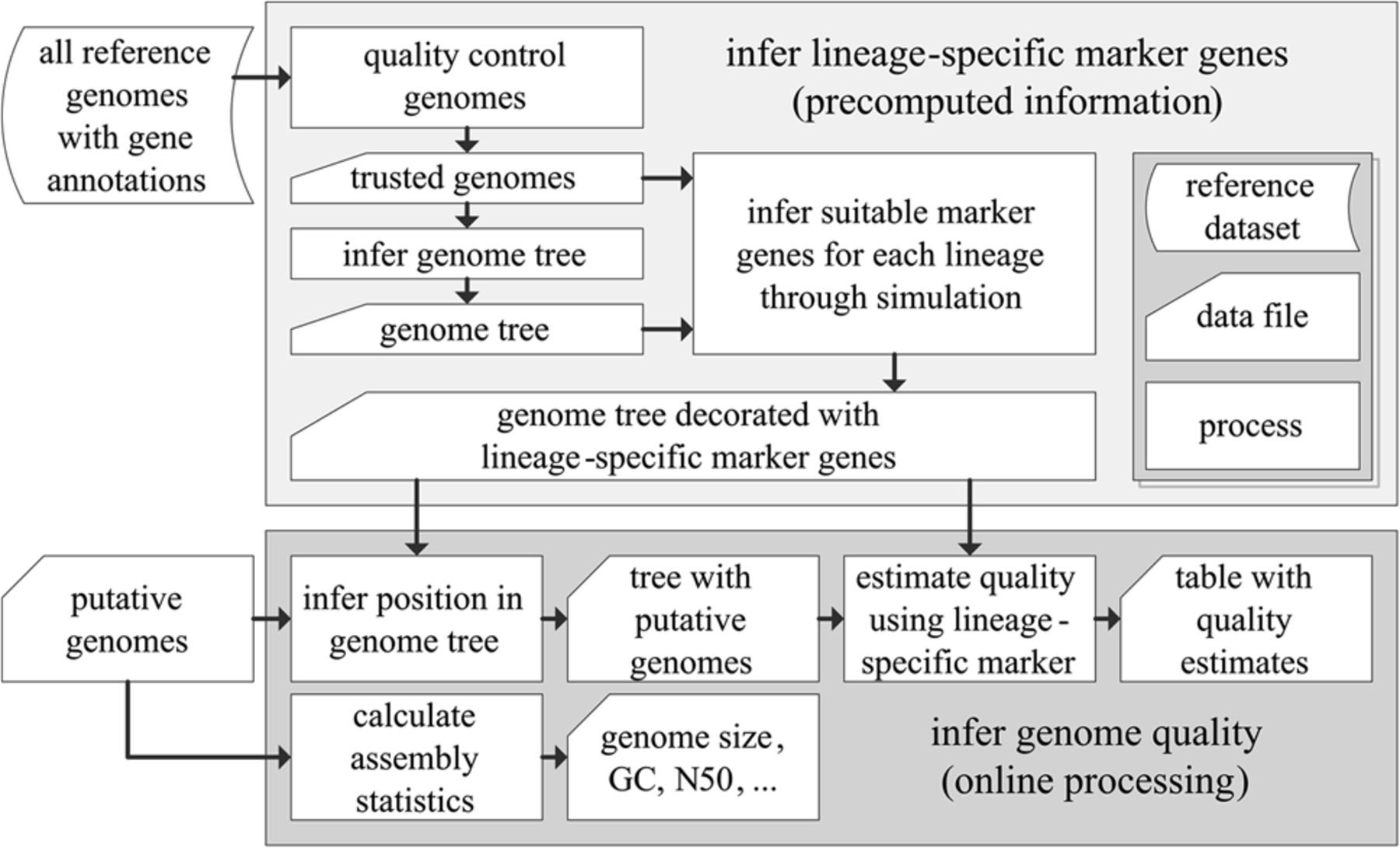
CheckM consists of a workflow for precomputing lineage-specific marker genes for each branch within a reference genome tree (top box) and an online workflow for inferring the quality of putative genomes (bottom box). Starting with a set of annotated reference genomes, the quality of these genomes is assessed in order to produce a set of near-complete genomes suitable for inferring marker genes. These genomes form the basis of a reference genome tree. A simulation framework is then used to associate each branch in the reference genome tree with a lineage-specific marker set suitable for robustly estimating the quality of genomes placed along a given branch (Fig. 3). To determine the quality of a putative genome, its position within the reference genome tree is inferred in order to establish the set of marker genes suitable for assessing its quality. These marker genes are identified within the putative genome and the presence/absence of these genes used to estimate its completeness and contamination.








