
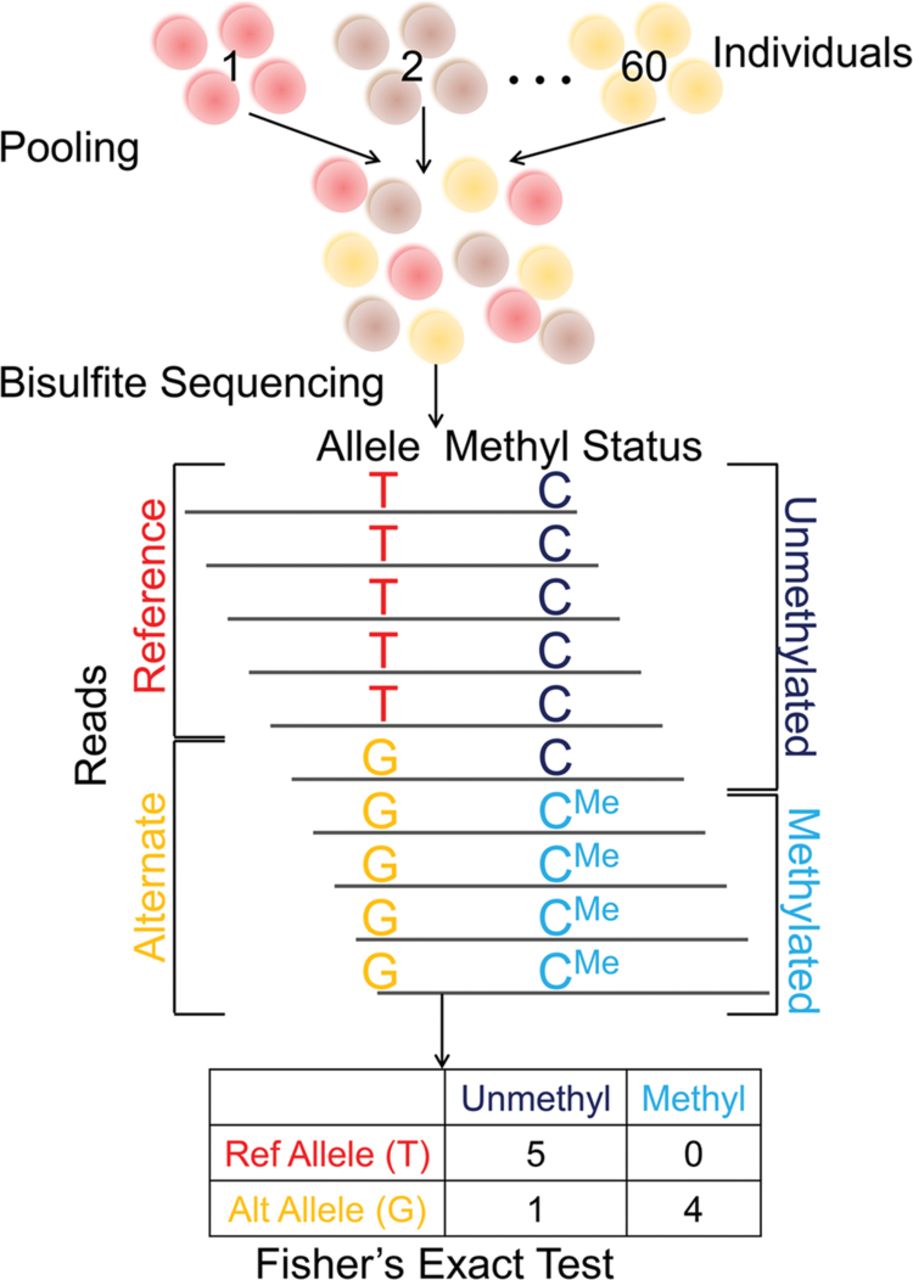
Figure 1.
We use pooled bisulfite sequencing to identify mQTLs. Our pooling method enables us to identify mQTLs directly from bisulfite sequencing reads. In this approach, cells or DNA samples from all individuals are combined into a single pool, which is then subject to bisulfite sequencing. Alleles and methylation statuses are inferred from the sequence reads, which are then used to generate a 2 × 2 contingency table (where columns represent methylation statuses and rows represent alleles). Fisher's exact test is used to compute a P-value for the null hypothesis of no association.








