
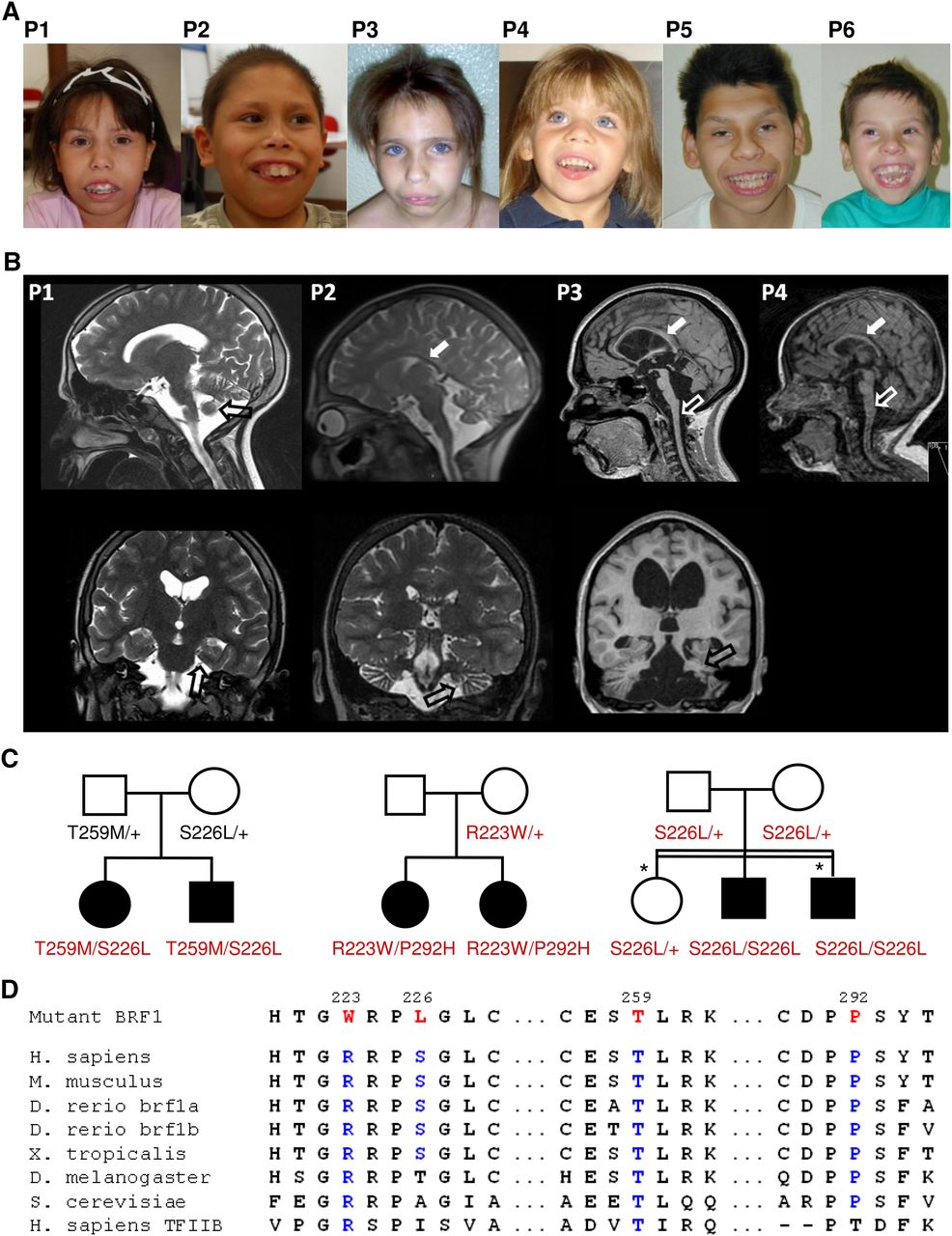
BRF1 mutations cause a cerebellar-dental-skeletal syndrome. (A) Patients 1 and 2 (family 1) at the ages of 12 and 10 yr, patients 3 and 4 (family 2) at the ages of 10 and 4 yr, and patients 5 and 6 (family 3). Note characteristic facial dysmorphism and dental anomalies. (B) Brain MRI (top, sagittal scans; bottom, coronal scans) of patients 1–4 (P1–P4) at the ages of 14 yr, 9 yr, 12 yr, and 18 mo, respectively, showing a thin corpus callosum (white filled arrows), flattened brainstem (white unfilled arrows), and cerebellar hypoplasia (black unfilled arrows). (C) Pedigrees of family 1 (left), family 2 (middle), and family 3 (right) with genotypes for BRF1 missense alterations. In family 2, the p.Pro292His mutation was likely transmitted by the unaffected father who did not participate in the study. In family 3, the two individuals denoted by an asterisk had Leber congenital amaurosis caused by a homozygous RDH12 mutation. (D) Multiple sequence alignment of BRF1 orthologs and human TFIIB showing evolutionary conservation of mutant BRF1 amino acid residues.








