
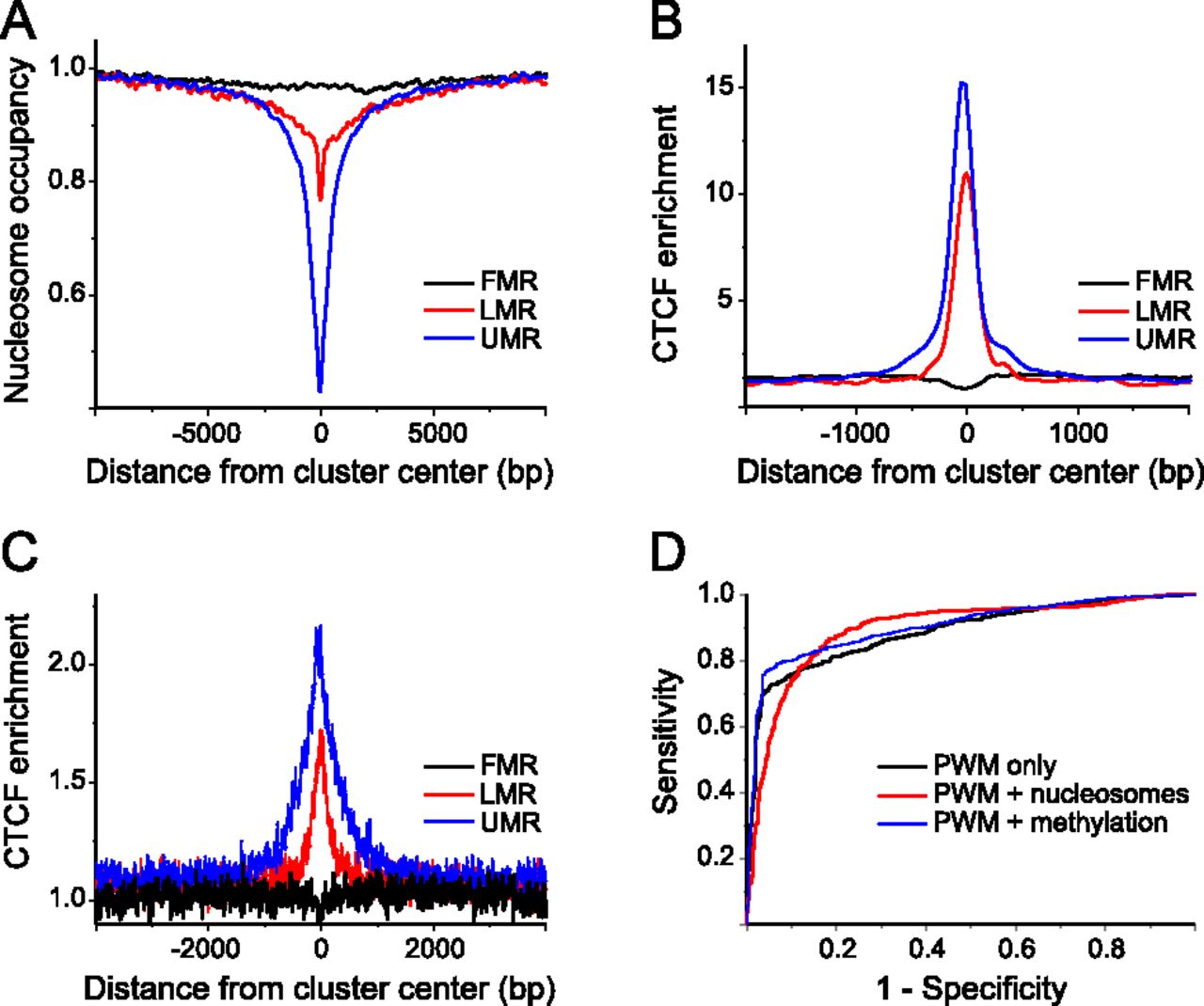
CTCF binding in ESCs can be explained solely by the DNA sequence. (A) Average nucleosome occupancy profiles for genomic regions with different levels of DNA methylation. For fully methylated regions (FMR), the occupancy remains flat as compared to genome-average levels. Low methylated regions (LMR) and unmethylated regions (UMR) were nucleosome-depleted by ∼30% and ∼60%, respectively. (B) Average CTCF enrichment calculated from ChIP-seq data for the three different classes of 5mC density. (C) Average CTCF enrichment predicted for the same regions based on the DNA sequence preferences given by the TRANSFAC PWM, without taking into account nucleosomes and DNA methylation. (D) Receiver operator curves calculated for the TFnuc model, taking into account only the CTCF weight matrix without nucleosomes and DNA methylation (black). In addition to PWM, competition with nucleosomes (red) or DNA methylation (blue) was considered. The area under the curve (AUC) reflects both the sensitivity and specificity and thus determines the goodness of the model.








