
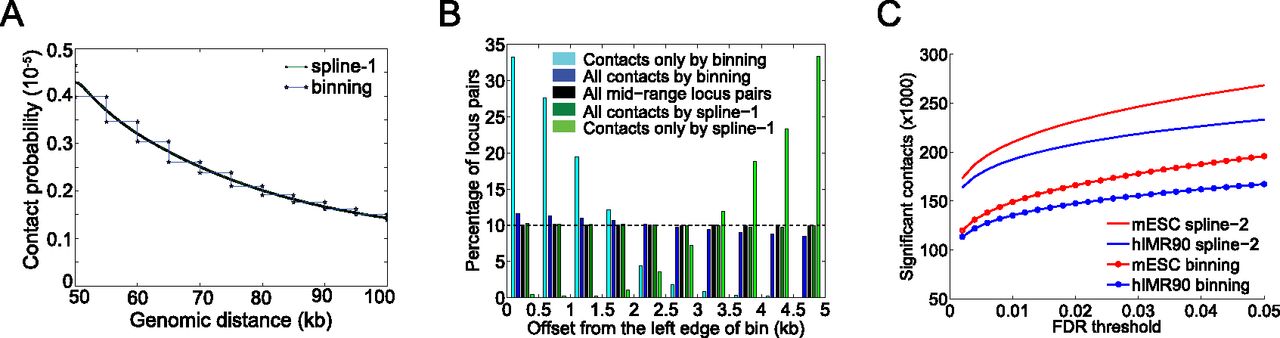
Fit-Hi-C eliminates edge effects caused by discrete binning and boosts statistical power for confidence estimation. (A) Comparison of fits resulting from our method to the discrete binning method which uses 5-kb genomic distance bins for a S. cerevisiae HindIII library from Duan et al. (2010). Only the genomic distance range of 50–100 kb is shown for visualization purposes. (B) Histograms of genomic distance offsets (Methods) for contacts identified at FDR 1% from ICE-corrected contact maps by different methods (dark blue, dark green) and for all possible mid-range locus pairs (black) for the library described in A. Histograms for each series sums up to 100%. For each locus pair, the offset is calculated with respect to the left edge of the enclosing 5-kb bin. Because we divide each 5-kb bin into 10 equally sized windows, we expect a contact set with no binning bias to have 10% (dashed line) of its members in each of these 10 windows, similar to the set of all mid-range locus pairs (black). We quantify the bias by testing the null hypothesis that the proportion of significant contacts on each side of the bin identified by a specific method is equal to this proportion for all mid-range locus pairs (black). Fisher's exact test P-values for this null hypothesis are 8.3 × 10−44 and 0.34 for all contacts identified by binning (dark blue) and by spline-1 (dark green), respectively. (C) Comparison of the number of contacts deemed significant by the refined null-based spline fit (spline-2) and discrete binning methods at varying FDR thresholds for hIMR90 and mESC libraries from Dixon et al. (2012).








