

Outline of the Integrated System for Motif Activity Response Analysis. (A) ISMARA starts from a curated genome-wide collection of promoters and their associated transcripts. Using a comparative genomic
Bayesian methodology (Arnold et al. 2012a), transcription factor binding sites (TFBSs) for ∼200 regulatory motifs are predicted in proximal promoters. Similarly, miRNA
target sites for ∼100 seed families are annotated in the 3′ UTRs of transcripts associated with each promoter (Friedman et al. 2009). (B) Users provide measurements of gene expression (microarray, RNA-seq) or chromatin state (ChIP-seq). The raw data are processed
automatically, and a signal is calculated for each promoter in each sample. For ChIP-seq data, the signal is calculated from
the read density in a region around the transcription start. For gene expression data, the signal is calculated from read
densities across the associated transcripts (RNA-seq) or intensities of associated probes (microarray). (C) The site predictions and measured signals are summarized in two large matrices. The components Npm of matrix N contain the total number of sites for motif m (TF or miRNA) associated with promoter p. The components Eps of matrix E contain the signal associated with promoter p in sample s. (D) The linear MARA model is used to explain the signal levels Eps in terms of bindings sites Npm and unknown motif activities Ams, which are inferred by the model. The constants cp and 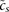 correspond to basal levels for each promoter and sample, respectively. (E) As output, ISMARA provides the inferred motif activity profiles Ams of all motifs across the samples, s, sorted by the significance of the motifs. A sorted list of all predicted target promoters is provided for each motif, together
with the network of known interactions between these targets (provided by the String database, http://string-db.org/) and a list of Gene Ontology categories that are enriched among the predicted targets. Finally, for each motif, a local network
of predicted direct regulatory interactions with other regulators is provided.
correspond to basal levels for each promoter and sample, respectively. (E) As output, ISMARA provides the inferred motif activity profiles Ams of all motifs across the samples, s, sorted by the significance of the motifs. A sorted list of all predicted target promoters is provided for each motif, together
with the network of known interactions between these targets (provided by the String database, http://string-db.org/) and a list of Gene Ontology categories that are enriched among the predicted targets. Finally, for each motif, a local network
of predicted direct regulatory interactions with other regulators is provided.








