
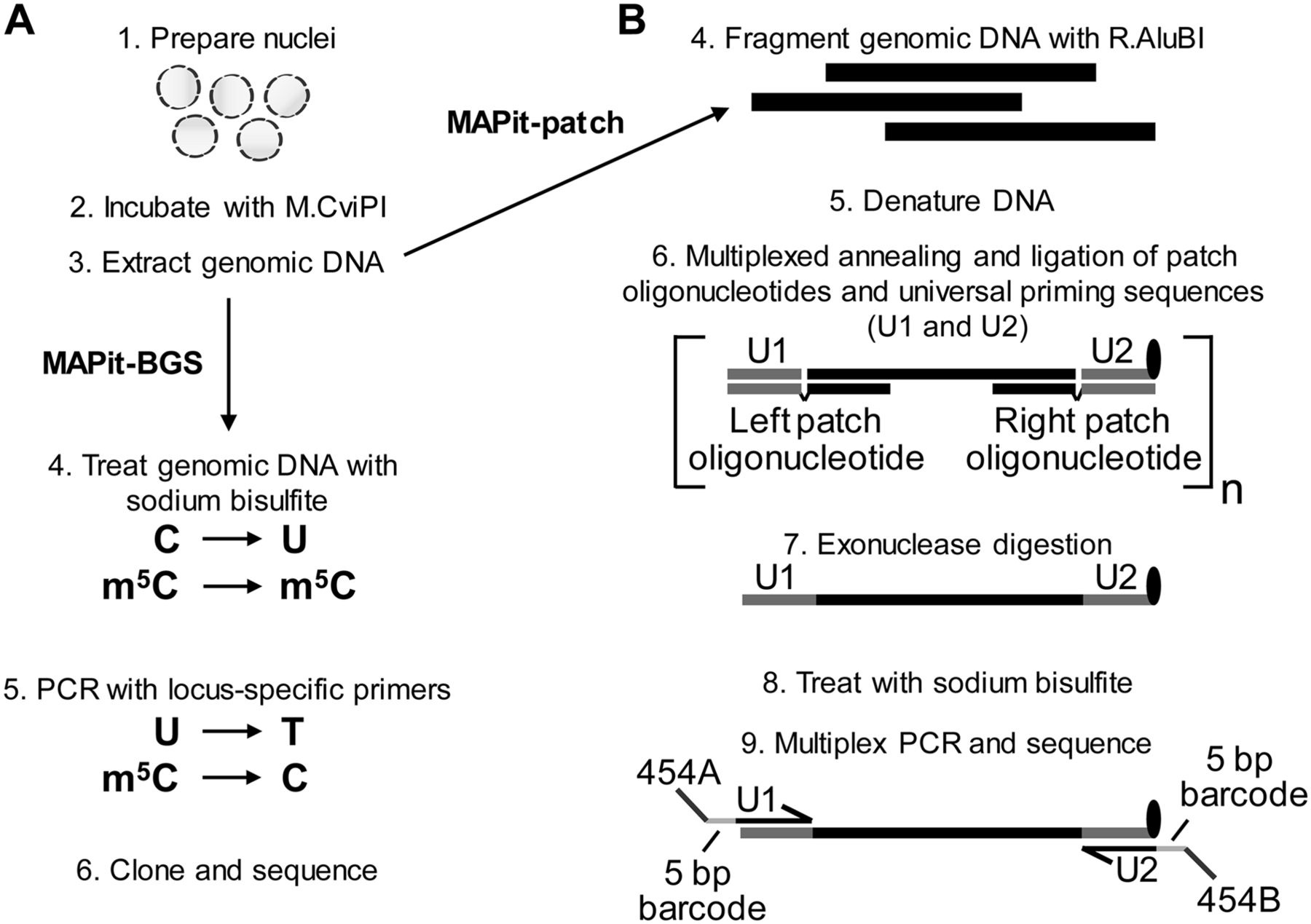
MAPit-BGS and MAPit-patch workflow. Both assays begin with (1) preparation of nuclei and (2) incubation with M.CviPI. Upon termination of the chromatin probing reaction, (3) genomic DNA is extracted and processed. (A) For MAPit-BGS, genomic DNA is (4) bisulfite treated such that unmethylated C is deaminated to U, whereas methylated C (m5C) remains m5C. Bisulfite-treated DNA is then (5) PCR amplified using locus-specific primers; then reaction products are (6) purified and cloned. Individual clones are sequenced and data are analyzed to map the methylation status of CG and GC sites. (B) For MAPit-patch, (4) genomic DNA is fragmented using a GC and CG methylation-insensitive enzyme, such as R.AluBI. Fragmented DNA is then (5) denatured and (6) subjected to target selection, whereby left and right patch oligonucleotides hybridize to each end of one strand of each locus and “patch” complementary oligonucleotides for universal priming (U1 and U2) by ligation (step not shown). The U2 oligonucleotide contains exonuclease-resistant modifications at its 3′ end (black oval). Therefore, subsequent (7) 3′ to 5′ exonuclease digestion leaves targeted DNA strands intact and removes unhybridized oligonucleotides as well as nontargeted genomic DNA. Enriched DNA is (8) bisulfite converted and (9) amplified using universal primers that comprise sequences of U1 or complementary to U2, 5-bp barcodes to facilitate multiplexing, and adapter sequences specific for a sequencing platform.








