

Correction to read counts. Copy number profiles from (A) uncorrected and (C) corrected read counts; (B) median read counts per bin as a function of GC content and mappability; and (D) the corresponding LOESS fit for sample LGG150. Regions of the isobar plots that are white contain no bins with that combination
of GC and mappability. In the copy number profiles, bins are ordered along the x-axis by their genomic positions, and the y-axis shows median-normalized log2-transformed data. Small triangles at the top and bottom edges represent data points that fall outside the plot area. Upper left corners show the number and size of bins. Upper right corners of the median read counts plot shows the total number of sequence reads, and upper right corners of the copy number profiles the expected and measured standard deviation. The expected standard deviation (E σ) is defined as 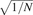 , where N is the average number of reads per bin. The measured standard deviation
, where N is the average number of reads per bin. The measured standard deviation 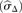 is calculated from the data with a mean-scaled and 0.1%-trimmed first-order estimate, prior to log2 transforming the data for plotting (see text).
is calculated from the data with a mean-scaled and 0.1%-trimmed first-order estimate, prior to log2 transforming the data for plotting (see text).








