
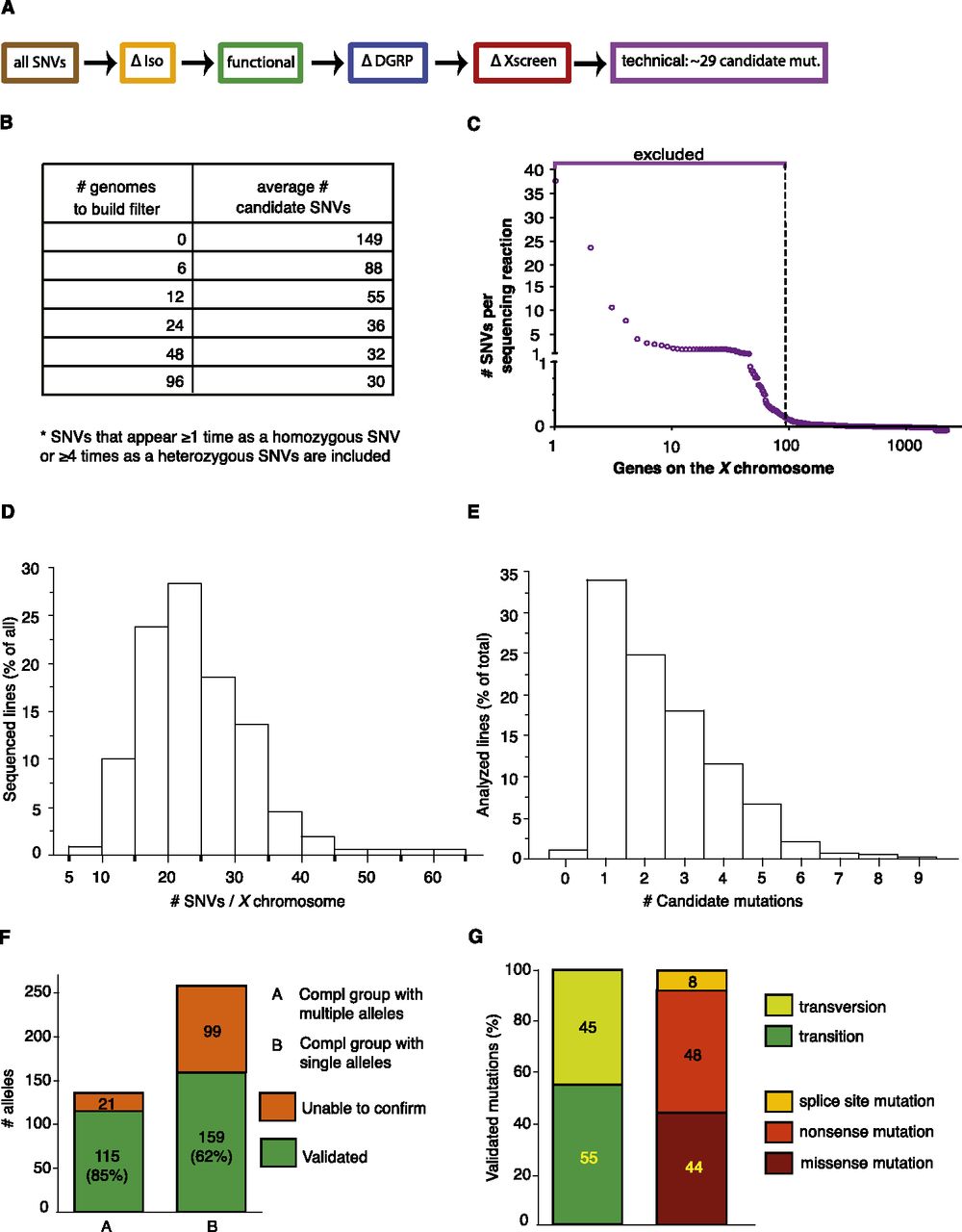
Filtering strategy to identify candidate genes in transheterozygous (mut 1/mut 2) mutants. (A) The same filters were applied as in Figure 2, and additional filters were added to remove SNVs identified repeatedly in multiple sequenced genomes (ΔXscreen [red]). A final filter was added to exclude genes that appear to be difficult to sequence (technical [purple]). (B) Building a background-specific filter (∆Xscreen). The largest drop in SNVs is seen when the ΔXscreen filter is built based on recurring SNVs found in 12 transheterozygous mutant genomes. (C) Building a technique-specific filter (technical). Approximately 95 genes appear difficult to sequence or analyze, since SNVs in these genes are called in nearly every sequenced genome. Hence, these genes were excluded from analysis (see Supplemental Table 1). (D) Distribution of the number of SNVs per chromosome that were identified in all analyzed sequence files. On average, 15 to 25 SNVs were identified for the two X chromosomes sequenced in the same reaction. (E) Distribution of the number of identified candidate mutations in an ∼1.4-Mb region to which lethality was mapped by duplication mapping. On average, one to two candidate mutations were found per duplication. (F) Mapping efficiency. For complementation groups consisting of multiple alleles, the causative mutation could be identified in 85% of the sequenced lines, as they could be rescued by an 80-kb P[acman] construct. For single alleles, the mutation could be validated in 62% of the sequenced lines. (G) Characteristics of the identified mutations.








