
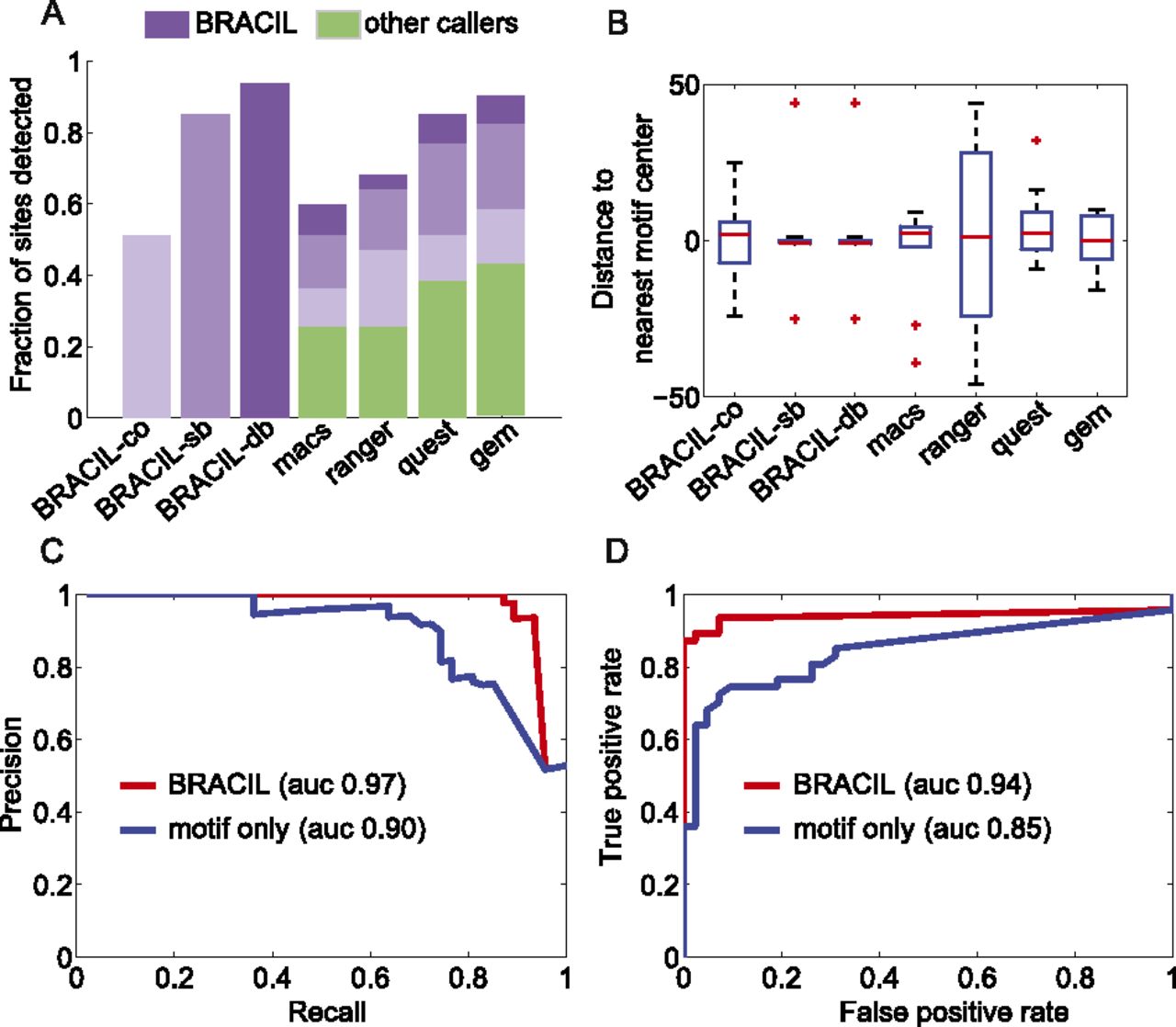
BRACIL increases the resolution of binding-site detection as well as sensitivity and specificity. The performance achieved by using only ChIP-seq coverage (BRACIL-co) is improved by including motif integration with the single-binding signal (BRACIL-sb), and the best performance occurs when we also consider the double-binding signal (BRACIL-db). A summary of the differences between the three versions can be found in Table 1. In the best scenario, BRACIL detects 44/47 of the reference sites (Chauhan et al. 2011). The potential of our method to refine the output of peak callers can be seen both in terms of the fraction of sites detected (A) as well as the resolution with which they are detected (B). The green bars (A) represent the fraction of sites detected by the corresponding peak caller labeled at the x-axis. The purple bars on the top show the additional refinement provided by our method. The different shades in purple represent performance improvement by specific variations of BRACIL. Enriched regions are defined as the overlapping window of ±150 bp around the single nucleotide prediction obtained by the corresponding peak caller. For BRACIL-co, BRACIL-sb, and BRACIL-db, the enriched regions consist in the ±150-bp overlapping window that surrounds the reference sites. The precision and recall (C) and the ROC (D) plots illustrate that our method is especially important for filtering out low conservation motifs that do not represent real binding.








