
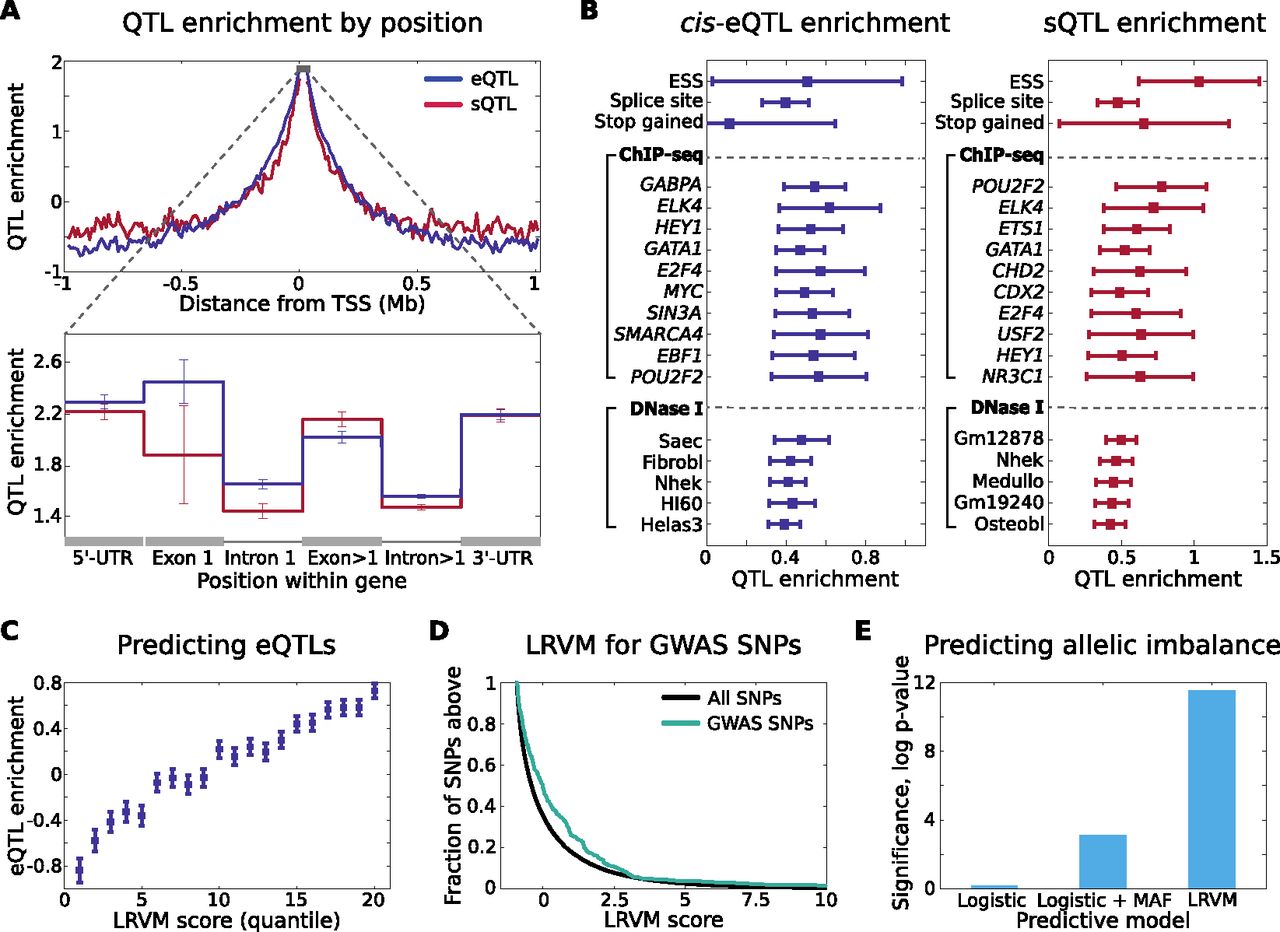
Genomic properties of regulatory variation and prediction of eQTLs. (A) Enrichment of proximal eQTLs and sQTLs is shown as a function of distance to the TSS. Enrichment is computed here as the log odds multiplier on likelihood of association (Methods). In the zoomed, intrageneic view, enrichment (log odds multiplier) of proximal eQTLs and sQTLs is shown within gene boundaries for UTR, intronic, and exonic loci. We aggregate SNPs within all exons except the first (the closest to TSS) together, and likewise for introns. (B) Enrichment of cis-eQTLs and sQTLs for functional and genomic annotations, controlling for distance. In each case, (log) odds multipliers were computed for each category after conditioning on SNP location (Methods) shown in A and B. ChIP-seq and DNase I annotation enrichments are shown here for SNPs falling within 20 kb upstream of TSS; for full enrichment statistics see Supplemental Data S2. (C) Enrichment of cis-eQTLs stratified by LRVM score (restricting to genes and SNPs excluded from training LRVM). Each SNP-gene pair was scored by LRVM for likelihood of association, and twenty quantiles were computed for the resulting scores. Finally, enrichment was computed for each quantile, using log odds estimation after correcting for position (Methods). (D) Predicted regulatory impact of trait-associated (GWAS) SNPs according to LRVM for 263 unique disease variants not available during LRVM training. We compute the score of each SNP for each of its proximal genes. Known trait-associated SNPs score more highly that expected at random (P < 10−9), indicating enrichment for properties that match those of observed regulatory variants. (E) LRVM scores are predictive of allelic effects, indicative of cis-regulatory impact. We correlate allelic imbalance (Methods) observed among heterozygous individuals for each SNP with the score assigned by each predictive model to the corresponding SNP. Significance is estimated using the Wilcoxon rank sum test. Again, analysis is restricted to SNPs not used to train LRVM.








