
Small noncoding differentially methylated copy-number variants, including lncRNA genes, cause a lethal lung developmental disorder
- Przemyslaw Szafranski1,
- Avinash V. Dharmadhikari1,2,
- Erwin Brosens3,4,
- Priyatansh Gurha1,5,
- Katarzyna E. Kołodziejska1,
- Ou Zhishuo1,
- Piotr Dittwald1,6,7,
- Tadeusz Majewski8,
- K. Naga Mohan1,9,
- Bo Chen1,
- Richard E. Person1,
- Dick Tibboel4,
- Annelies de Klein3,
- Jason Pinner10,
- Maya Chopra10,
- Girvan Malcolm11,
- Gregory Peters12,
- Susan Arbuckle13,
- Sixto F. Guiang III14,
- Virginia A. Hustead15,
- Jose Jessurun16,
- Russel Hirsch17,
- David P. Witte17,
- Isabelle Maystadt18,
- Neil Sebire19,
- Richard Fisher20,
- Claire Langston21,
- Partha Sen22 and
- Paweł Stankiewicz1,2,23
- 1Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas 77030, USA;
- 2Program in Translational Biology and Molecular Medicine, Baylor College of Medicine, Houston, Texas 77030, USA;
- 3Clinical Genetics Department, Erasmus MC-Sophia, 3000 CA, Rotterdam, Netherlands;
- 4Paediatric Surgery, Erasmus MC-Sophia, 3015 GJ, Rotterdam, Netherlands;
- 5Center for Cardiovascular Genetics, The Brown Foundation Institute of Molecular Medicine, The University of Texas Health Science Center and Texas Heart Institute, Houston, Texas 77030, USA;
- 6Institute of Informatics, University of Warsaw, 02-097, Warsaw, Poland;
- 7College of Inter-Faculty Individual Studies in Mathematics and Natural Sciences, University of Warsaw, 02-089, Warsaw, Poland;
- 8Department of Pathology, University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA;
- 9Department of Biological Sciences, Birla Institute of Technology and Science-Pilani, Hyderabad-500 078, AP, India;
- 10Department of Molecular and Clinical Genetics, Royal Prince Alfred Hospital, Sydney, NSW 2050, Australia;
- 11Obstetrics, Gynaecology and Neonatology, Royal Prince Alfred Hospital, Sydney, NSW 2006, Australia;
- 12Cytogenetics Department, The Children's Hospital at Westmead, Westmead, NSW 2145, Australia;
- 13Histopathology Department, The Children's Hospital at Westmead, Westmead, NSW 2145, Australia;
- 14Department of Pediatrics–Neonatology, University of Minnesota Medical Center, Minneapolis, Minnesota 55454, USA;
- 15Minnesota Neonatal Physicians, Minneapolis, Minnesota 55404, USA;
- 16Laboratory Medicine and Pathology, Department of Medicine, University of Minnesota Medical Center, Minneapolis, Minnesota 55454, USA;
- 17Cincinnati Children's Hospital Medical Center, Cincinnati, Ohio 45229, USA;
- 18Institut de Pathologie et de Génétique, B-6041 Gosselies, Belgium;
- 19Department of Paediatric Histopathology, Great Ormond Street Hospital for Children and UCL Institute of Child Health, London WC1N 3JH, United Kingdom;
- 20James Cook University Hospital, Middlesborough TS4 3BW, United Kingdom;
- 21Department of Pathology, Baylor College of Medicine, and Pulmonary Pathology, Texas Children's Hospital, Houston, Texas 77030, USA;
- 22Department of Pediatrics–Neonatology, Baylor College of Medicine, Houston, Texas 77030, USA
Abstract
An unanticipated and tremendous amount of the noncoding sequence of the human genome is transcribed. Long noncoding RNAs (lncRNAs) constitute a significant fraction of non-protein-coding transcripts; however, their functions remain enigmatic. We demonstrate that deletions of a small noncoding differentially methylated region at 16q24.1, including lncRNA genes, cause a lethal lung developmental disorder, alveolar capillary dysplasia with misalignment of pulmonary veins (ACD/MPV), with parent-of-origin effects. We identify overlapping deletions 250 kb upstream of FOXF1 in nine patients with ACD/MPV that arose de novo specifically on the maternally inherited chromosome and delete lung-specific lncRNA genes. These deletions define a distant cis-regulatory region that harbors, besides lncRNA genes, also a differentially methylated CpG island, binds GLI2 depending on the methylation status of this CpG island, and physically interacts with and up-regulates the FOXF1 promoter. We suggest that lung-transcribed 16q24.1 lncRNAs may contribute to long-range regulation of FOXF1 by GLI2 and other transcription factors. Perturbation of lncRNA-mediated chromatin interactions may, in general, be responsible for position effect phenomena and potentially cause many disorders of human development.
Mammalian regulatory elements, including enhancers, repressors, and insulators, are usually contained in genomic regions that lack protein-coding genes and can be located hundreds of kilobases from genes that they regulate (Birney et al. 2007). High-throughput analysis of the human transcriptome revealed that the majority of the noncoding portion of the genome is transcribed as regulatory RNAs (Khalil et al. 2009; Mattick 2010). Long noncoding RNAs (lncRNAs) are non-protein-coding transcripts longer than 200 nt, distinguished from small regulatory RNAs such as microRNAs, siRNAs, piRNAs, and snoRNAs. Little is known about the function of lncRNAs in humans with XIST being the earliest identified and best characterized to date human lncRNA. Recently, lncRNAs have been implicated in imprinting and long-range gene regulation with functional properties of enhancers (Khalil et al. 2009; Mattick 2010; Wang and Chang 2011). Here, we demonstrate that loss of a distant enhancer region in a protein-coding gene desert at 16q24.1, which includes lung-expressed lncRNA genes, leads to alveolar capillary dysplasia with misalignment of pulmonary veins (ACD/MPV; MIM 265380).
ACD/MPV is a rare, neonatally lethal developmental disorder of the lungs defined by malposition of pulmonary veins adjacent to small pulmonary arteries, medial thickening of small pulmonary arteries, deficient lobular development, a paucity of alveolar wall capillaries, and occasional lymphangiectasis (Langston 1991; Bishop et al. 2011). A few familial ACD/MPV cases have been described (Sen et al. 2004, 2012; Eulmesekian et al. 2005). During the last 10 years, we ascertained ACD/MPV samples (mainly formalin-fixed paraffin-embedded lung tissues) from more than 90 families. In 2009, we reported genomic deletions and inactivating point mutations in the FOXF1 gene in chromosome 16q24.1 in unrelated patients with sporadic histopathologically verified ACD/MPV (Stankiewicz et al. 2009).
FOXF1 (Forkhead box protein F1) is a member of the FOX transcription factor family sharing a winged helix/forkhead DNA-binding domain (Lai et al. 1993; Pierrou et al. 1994; Kaestner et al. 2000). FOXF1 and its mouse ortholog are predominantly expressed in subepithelial mesenchymal tissues of developing lung and foregut (Chang and Ho 2001; Mahlapuu et al. 2001a; Maeda et al. 2007; Bozyk et al. 2011). Homozygous mice deficient for Foxf1 die in utero by embryonic day 10 (E10) due to defects in mesodermal differentiation and cell adhesion (Mahlapuu et al. 2001b). Interestingly, ∼50% of Foxf1+/− mice died from pulmonary hemorrhage and showed severe defects in alveolarization and vasculogenesis (Kalinichenko et al. 2001), partially recapitulating histopathological pulmonary defects in infants with ACD/MPV, whereas the remainder appeared normal.
Both in vitro and in vivo studies in mice have shown that none of the in silico–predicted transcription factor binding sites located within the Foxf1 promoter confer its tissue specificity (Chang and Ho 2001; Kim et al. 2005), suggesting that the promoter is regulated by tissue-specific distant regulatory elements. Also, deletions upstream of FOXF1, leaving FOXF1 intact, have been described in two patients with ACD/MPV, suggesting the presence of distant regulatory elements for FOXF1 (Stankiewicz et al. 2009). In addition, FOXF1 has been bioinformatically predicted to be paternally imprinted (Luedi et al. 2007), and other studies have supported this prediction (Stankiewicz et al. 2009; Sen et al. 2012).
We now report novel overlapping de novo noncoding deletion copy-number variants (CNVs) located 96–257 kb upstream of FOXF1 in seven patients with ACD/MPV, who all died in the first month of life with severe respiratory distress and pulmonary hypertension. Based on extensive experimental characterization of the microdeletion-defined upstream regulatory region, we propose that paternally incompletely imprinted FOXF1 is regulated in cis by an interplay between chromatin looping, possibly with contribution of lncRNAs, and methylation-controlled GLI2 binding.
Results
Noncoding deletion CNVs upstream of FOXF1
DNA samples from nine patients with ACD/MPV, negative for point mutations within the FOXF1 coding exons, were analyzed by array comparative genomic hybridization (aCGH) for the presence of CNVs in 16q24.1. In sample 59.4, aCGH revealed a 15-kb deletion of the entire FOXF1 and its promoter. Studies of the remaining eight cases identified CNVs that left the FOXF1 coding region intact. In patient 41.4, an ∼11-kb deletion, mapping 2.6 kb upstream of FOXF1, removed one copy of the distal portion of the FOXF1 promoter, corresponding to Foxf1 promoter region II in mice (Kim et al. 2005) and overlapping with a portion of the lncRNA gene FOXF1-AS1 (also known as lncFOXF1 [Khalil et al. 2009] and TCONS_00024240) (Ota et al. 2004). We show by RT-PCR and DNA sequencing that FOXF1-AS1 is expressed in normal human newborn lungs (Supplemental Fig. S1). In the remaining seven patients, overlapping deletions, ranging in size from 140 kb to 2625 kb, mapped 96–257 kb upstream of FOXF1 (Fig. 1; Supplemental Table S1).
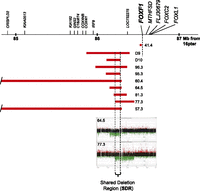
Nine (two published—D9, D10—and seven novel) ACD/MPV-causing microdeletions share a 75-kb putative regulatory region (SDR), mapping 257 kb upstream of FOXF1 (16q24.1). Array CGH plots of two deletions defining SDR are shown.
To better estimate the recurrence risk for these deletions, we mapped and sequenced the deletion breakpoints of six of the upstream microdeletions (patients 57.3, 60.4, 64.5, 77.3, 95.3, and 96.3) (Supplemental Fig. S2; Supplemental Table S1) and the microdeletion that included the FOXF1 gene (patient 59.4) (Supplemental Fig. S2). Microhomology was identified in five of seven breakpoint junctions (patients 57.3, 60.4, 64.5, 77.3, and 95.3), consistent with the deletions arising by a template switching replicative mechanism such as FoSTeS/MMBIR (Lee et al. 2007; Hastings et al. 2009). In three of these five cases, both deletion breakpoints mapped within Alu elements (Supplemental Table S1). The probability of finding both breakpoints in Alu elements by chance was estimated at less than 0.0066 (Supplemental Fig. S3), thus being much lower than the observed frequency of finding breakpoints of ACD/MPV microdeletions in Alu repetitive elements. This finding suggests a mechanistic link between Alu sequences and the location of microdeletion breakpoints. Interestingly, in patients 64.5 and 95.3, the distal breakpoints were located within the same copy of an AluSx element (∼100 bp apart), thus defining a potential breakage-prone hotspot. In patient 59.4, the breakpoints occurred within highly homologous regions of the low-complexity/GC-rich repeats and SINE/MIR repeats. We did not find any evidence for low-level somatic mosaicism in parental blood samples using PCR for patient-specific junction fragments; all microdeletions appear to have occurred de novo (Supplemental Fig. S4).
Regulatory region upstream of FOXF1
Sequence alignments of the seven distant microdeletions showed that they share a 75-kb region 257 kb upstream of FOXF1 (chr16:86,212,040–86,287,054; shared deletion region, SDR) (Fig. 1; Supplemental Table S1). We found that the SDR represents a protein–gene desert (http://genome.ucsc.edu). Nevertheless, this genomic region harbors DNA segments that are evolutionarily conserved among land vertebrates and have high 7× regulatory potential (Fig. 2A). Hence, we hypothesized that the SDR may contain regulatory site(s) controlling the FOXF1 promoter.
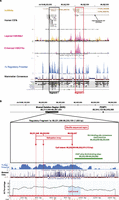
Characterization of the SDR. (A) Segment 1 (∼10 kb; black rectangle) and Segment 2 (∼8 kb; red rectangle), harboring, among others, transcription factor binding sites and a part of the fetal lung–expressed lncRNA gene, are shown. The figure is drawn according to the 2009 Human Reference Sequence (GRCh37/hg19). (B) Insight into the ∼1.5-kb Fragment 1a showing (from top to bottom) the bisulfite sequenced region, methylation array oligo probes (red arrows) that indicate differential methylation, differentially methylated CpG island, ChIP-chip GLI2-binding region, 7× regulatory potential, mammalian evolutionary conservation, and CG content.
Interestingly, we also found that the SDR encompasses one locus (chr16:86,223,827–86,234,547) and part of another locus (chr16:86,254,429–86,338,058), encoding differentially spliced putative lncRNAs exhibiting increased expression in the lungs (http://genome.ucsc.edu). The 43.5-kb EST CR737045 (chr16:86,259,186–86,302,689), part of the 79-kb lncRNA TCONS_00024764, is specifically expressed in fetal lungs (SOURCE at http://source.stanford.edu). We show, by RT-PCR and sequencing, that TCONS_00024764 lncRNA gene is also expressed in normal newborn lung at a much higher level than in HEK293 kidney cells and lymphoblasts (Supplemental Fig. S1).
We further narrowed SDR to ∼10-kb Segment 1 of the highest 7× regulatory potential and sequence conservation (Fig. 2A), containing a cluster of the predicted binding sites for the GLI1, GLI2, and GLI3 transcription factors (GLI-binding sites, consensus sequence: GACCACCCACNNNG [Hallikas et al. 2006]) overlapping with a CpG island (Fragment 1a) (Fig. 2B; Supplemental Fig. S5), and ∼8-kb Segment 2 with multiple binding sites for other transcription regulators, and the highest potential for histone methylation and acetylation. Both segments encode lncRNAs expressed in the lungs (Fig. 2A).
GLI2 binds within the upstream regulatory region
Sonic hedgehog (SHH) signaling targets a GLI2 transcription factor and is crucial for early lung development in mice (Rutter et al. 2010) and humans (Zhang et al. 2010). GLI1-3 have been shown to be expressed in human lung mesenchymal tissues, including endothelial cells (Maeda et al. 2007). Moreover, studies in mice have shown that Gli2 regulates Foxf1 expression in the developing stomach and intestine (Madison et al. 2009), which, together with airway and alveolar tissues, are of endodermal origin. To determine whether in silico–identified GLI-binding sites in the SDR bind GLI2 in vivo, we screened the SDR for the GLI2 binding using a ChIP-chip assay. We detected strong binding of GLI2 to SDR in the cultured human pulmonary microvascular endothelial cells (HPMEC) (p = 0.007, n = 2) at a region including several in silico–predicted GLI-binding sites (chr16:86,232,498–86,232,882) (Supplemental Figs. S5, S6). Although only one of the GLI-binding sites of SDR perfectly matches the consensus sequence, the clustering of nine of these sites within a 0.2-kb portion of Fragment 1a may have a synergistic effect on GLI2 binding. Given that HPMEC expresses both FOXF1 and GLI2 (Supplemental Fig. S7), we hypothesized that GLI2 binding at SDR may regulate FOXF1 transcription.
GLI2 binding to the distant enhancer regulates FOXF1 promoter
To this end, we performed a secreted alkaline phosphatase (SEAP) reporter assay in HPMEC (Fig. 3A,B). We prepared plasmid constructs that allow for transient expression of the SEAP reporter gene under the control of the FOXF1 promoter and its putative regulatory sites. The upstream putative regulatory sequences were then tested both in cis (inserted into a vector upstream of the FOXF1 promoter) and in trans (placed on another plasmid used in the cotransfection of HPMEC with the FOXF1 promoter–containing reporter vector). The −5.5-kb FOXF1 promoter region includes two intervals that remain highly conserved between mice and humans (Kim et al. 2005) and contains, besides the RNA Pol II–binding site (chr16:86,543,198–86,543,374), multiple transcription factor binding sites (http://genome.ucsc.edu). When placed upstream of the promoterless SEAP, the −5.5-kb promoter region activated SEAP transcription 11-fold to 20-fold; this range depended on whether the cells were additionally cotransfected with a pCS2Gli2 vector, constitutively expressing GLI2 (P < 0.0001) (Fig. 3A). Madison et al. (2009) demonstrated that the FOXF1 promoter harbors the evolutionarily conserved (identical among mammalian, bird, fish, and amphibian species) consensus GLI-binding site (chr16:86,539,377–86,539,385) that is deleted in patient 41.4 (Fig. 1). As expected, the truncated version of the FOXF1 promoter, missing this GLI-binding site, showed weaker SEAP expression in comparison with the intact promoter, especially in excess of GLI2 (P = 0.038) (Fig. 3A). Of note, several variant GLI-binding sites are still present in the residual truncated promoter region from case 41.4, potentially explaining why it responded, although weakly, to an excess of GLI2 (Fig. 3A). Placement of the 0.6-kb part of Fragment 1a of the SDR, containing GLI-binding sites, upstream of the FOXF1 promoter increased SEAP expression from the FOXF1 promoter twofold (P < 0.0016). Activation of the promoter by the SDR GLI-binding sites in trans also increased reporter expression (P = 0.027) (Fig. 3B), further suggesting that these GLI-binding sites can regulate FOXF1 transcription from a distant location, likely through chromatin looping that juxtaposes FOXF1 promoter and SDR. Moreover, cotransfection of HPMEC with the plasmid pCS2Gli2, constitutively expressing GLI2 protein, increased expression of the SEAP reporter from the FOXF1 promoter in all construct combinations that we tested (Fig. 3A,B), further indicating that FOXF1 can be regulated in lung endothelial cells by a GLI2 transcription factor. The increase of FOXF1 transcription from the −5.5-kb FOXF1 promoter alone, in response to the extra GLI2 synthesis, most likely results from binding of GLI2 to the mentioned consensus GLI-binding site or any of several GLI-binding site variants located within 5.5 kb upstream of FOXF1.
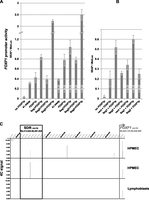
Distant regulatory region, SDR, controls the activity of the FOXF1 promoter. (A,B) Results of the reporter assay experiments showing regulation of the FOXF1 promoter in HPMEC by fragments of Segments 1 and 2 of the SDR. (FOXF1p) FOXF1 promoter (0–5.5 kb upstream of ATG codon) cloned in pSEAP2Basic; (41.4delp) truncated FOXF1 promoter (0 to −3.0 kb, ACD case 41.4) cloned in pSEAP2Basic; (Seg1FOXF1p) distant upstream GLI-binding region (part of Fragment 1a) and FOXF1 promoter cloned in pSEAP2Basic; (Seg2>FOXF1p) Segment 2 putative enhancer and FOXF1 promoter cloned in pSEAP2Basic in the same orientation; Seg2<FOXF1p, the same enhancer in reverse orientation and FOXF1 promoter cloned in pSEAP2Basic; (vect) pGEM-T Easy vector; (Seg1) GLI-binding region of Fragment 1a cloned in pGEM-T Easy; (Seg2) putative enhancer of Segment 2 cloned in pGEM-T Easy. The presence of additional GLI2, constitutively expressed from the pCS2Gli2 vector, is denoted by +GLI2. The GLI-binding region of Fragment 1a stimulates activity of the FOXF1 promoter in cis (A) and in trans (B) settings. (C) Results of the 4C experiment revealing long-range physical interaction between the FOXF1 promoter and SDR. Coordinates of the SDR region interacting with the FOXF1 promoter (SDR-promoter interaction peaks) in HPMEC are (from top) 86,246,725–86,246,982 (0.05 < FDR ≤ 0.1) and 86,246,125–86,246,982 (0.1 < FDR ≤ 0.2). The two HPMEC panels represent results of independent experiments. No SDR-FOXF1 promoter interaction peak was detected in lymphoblasts, which do not express FOXF1.
Using the SEAP reporter assay, we also tested an in silico–identified putative regulatory Segment 2, located ∼16 kb distally to the Segment 1 (Fig. 2A). This region was indicated by ChIP-seq assays to bind CTCF and several transcription factors other than GLI2 (http://genome.ucsc.edu). In the presence or absence of excess GLI2, a 1.4-kb fragment (chr16:86,256,619–86,258,038) of Segment 2 appeared to have no effect on the SEAP expression from the FOXF1 promoter when subcloned next to the FOXF1 promoter in the same orientation as the promoter, but increased the promoter activity fivefold when placed in the opposite orientation (P < 0.0001) (Fig. 3A). When tested in trans, the 1.4-kb fragment of Segment 2 did not significantly affect the FOXF1 promoter activity (Fig. 3B). Thus, in contrast to the GLI-binding sites of Fragment 1a, the tested fragment of Segment 2 seems unlikely to contribute to the long-range control of the FOXF1 expression.
SDR–FOXF1 promoter chromatin looping
\To determine whether there is a physical interaction between the FOXF1 promoter and the SDR that would juxtapose distant GLI-binding sites and the FOXF1 promoter, we performed a chromosome conformation capture-on-chip (4C) analysis. We detected interactions between the FOXF1 promoter region and regions located upstream of it, including the 75-kb SDR (Fig. 3C). Using two independently prepared 4C libraries from HPMEC, we identified overlapping SDR fragments (chr16:86,246,725–86,246,982 and 86,246,125–86,246,982) interacting with the FOXF1 promoter fragment (chr16:86,542,571–86,543,312) containing part of the Pol II–binding site that were not detected in the 4C library prepared from control lymphoblasts (Fig. 3C). These SDR fragments map 12 kb distal to Segment 1. Chromatin looping between SDR and the FOXF1 promoter could occur with a contribution of lncRNAs (Mattick 2009, 2010; Ørom et al. 2010; Wang and Chang 2011), including any of those encoded within SDR. Moreover, CTCF binding within the SDR (Supplemental Fig. S8) also might contribute to the formation of looping architecture (Phillips and Corces 2009). Since we did not detect interactions between SDR and the FOXF1 promoter in lymphoblasts, we propose that chromatin looping between SDR and the FOXF1 promoter (Supplemental Fig. S9) allows GLI2 to increase FOXF1 activity specifically in lung endothelium and other tissues of similar origin.
Maternal origin of ACD/MPV microdeletions
We have determined parental origin for seven out of eight upstream microdeletions (patients 57.3, 60.4, 64.5, 77.3, 81.3, 95.3, and 96.3) and for one microdeletion (59.4) that included the entire FOXF1. Interestingly, all these microdeletions arose de novo on the maternal chromosome (Supplemental Tables S1, S2). Six published de novo microdeletions that included FOXF1 (D1, D3, D4, and D8) or mapped upstream of FOXF1 (D9 and D10) also arose de novo on the maternal chromosome (Stankiewicz et al. 2009). Moreover, in a recently published unique familial case of ACD/MPV, with missense mutations in FOXF1 affecting five subjects, the mutation was inherited from a heterozygous healthy mother, in whom the mutation arose de novo on her paternal chromosome (Sen et al. 2012). In aggregate, these findings strongly suggest a parental-origin bias of FOXF1-associated CNVs (P < 0.001, n = 14).
Differential expression of FOXF1 parental alleles
To further test whether FOXF1 is imprinted, we compared FOXF1 expression levels from both parental chromosomes using two sets of semiquantitative RT-PCRs with primers designed to differentiate between the two parental chromosomes. We found that whereas both primer sets amplified equal amounts of FOXF1 from genomic DNA, their amplification from cDNA differed by 27% (P = 0.011, n = 3) (Supplemental Fig. S10). Thus, our data indicate that FOXF1 expression from the maternal chromosome is not equal to its expression from the paternal chromosome, further indicating that FOXF1 is imprinted in humans albeit incompletely.
Differentially methylated CpG site within SDR
Genomic imprinting can be caused by differential methylation of CpG islands, histone modification, or interactions with lncRNAs (Bartolomei 2009). Here, we have investigated in detail CpG methylation as one of the potential mechanisms of FOXF1 imprinting. The FOXF1 promoter resides within a large CpG island (Lo et al. 2010). We have bisulfite-sequenced this region of DNA from normal lung and have found that the FOXF1 promoter is not methylated (Fig. 4A). Previous studies using immortalized normal mammary cells and organoids have also shown that the FOXF1 promoter is not methylated (Lo et al. 2010). Interestingly, the part of the FOXF1 CpG island that is included in the FOXF1 first exon is extensively methylated (Fig. 4A); however, the functional significance of this methylation remains unknown. We also analyzed the results of the methylation studies using Infinium 450K methylation arrays on 39 brain DNA samples from normal individuals (courtesy of Dr. A.L. Beaudet). One of the 13 potentially differentially methylated regions upstream of FOXF1 encompassed three probes located within the SDR Fragment 1a in the vicinity of a cluster of GLI2-binding sites (Fig. 2B; Supplemental Fig. S5; Supplemental Table S3) identified in the ChIP-chip experiment and functionally verified in the reporter assay. Bisulfite sequencing of the SDR 0.2-kb CpG island closest to the consensus GLI2-binding site and overlapping with several GLI-binding site variants confirmed that it is differentially methylated in the DNA from normal fetal lung (Fig. 4A).
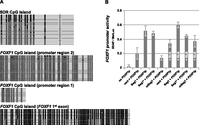
Methylation of the SDR CpG island decreases SDR potential to activate the FOXF1 promoter. (A) Methylation status of the FOXF1 promoter and SDR CpG island in normal fetal lung (a similar CpG methylation pattern was observed in DNA isolated from blood). Each lane represents a separate clone. (●) Methylated CpGs, (○) unmethylated CpGs. (Pink) Location of a cluster of GLI-binding site variants. (Red) Position of the consensus “core” GLI-binding site. Genomic coordinates of the bisulfite sequenced regions are: chr16:86,232,367–86,232,979 (SDR CpG island); chr16:86,542,223–86,542,807 (FOXF1 promoter 2); chr16:86,543,777–86,543,907 (FOXF1 promoter 1); chr16:6,544,458–86,545,037 (FOXF1 exon 1). (B) Dependence of the FOXF1 promoter activity on the methylation status of the SDR CpG island. The presence of additional GLI2 constitutively expressed from the pCS2Gli2 vector is denoted by +GLI2. (FOXF1p) FOXF1 promoter cloned in pSEAP2Basic; (vect) pGEM-T Easy; (Seg1) GLI-binding region (Fragment 1a) cloned in pGEM-T Easy; (mSeg1) GLI-binding region of Fragment 1a with in vitro–methylated CpG island cloned in pGEM-T Easy; (Seg1*) mock control for mSeg1.
Methylation of CpG island regulates GLI-binding sites
We were interested to know whether methylation of the CpG island, including and neighboring the GLI-binding sites, interferes with the ability of these sites to regulate FOXF1 expression. To this end, we have methylated (in vitro) half of the C residues of this CpG island in a 0.6-kb portion (chr16:86,232,261–86,232,908) of Fragment 1a. We then cotransfected the methylated construct with a FOXF1 promoter-harboring reporter plasmid. We observed a reduction in the ability of the Fragment 1a GLI-binding sites to activate the FOXF1 promoter, following partial methylation of the CpG island (P = 0.015) (Fig. 4B). The function of the methylated GLI-binding region did not significantly increase in the presence of excess GLI2 following the cotransfection of HPMEC with pCS2Gli2 (Fig. 4B).
Discussion
We identified overlapping deletions located upstream of FOXF1 in seven unrelated patients with ACD/MPV, defining a putative distant regulatory region (SDR) for FOXF1. This region, located 257 kb 5′ to FOXF1, is 75 kb in size and contains two smaller segments that are distinguished by high evolutionary conservation among land vertebrates and high 7× regulatory potential (Fig. 2). The ∼1.5-kb part of Segment 1 (Fragment 1a) encompasses several GLI-binding sites (Supplemental Fig. S5). GLI2 is essential for lung development in humans and mice (Maeda et al. 2007; Madison et al. 2009; Rutter et al. 2010; Zhang et al. 2010). Using a ChIP-chip assay, we have confirmed binding of GLI2 to Fragment 1a in HPMEC (Supplemental Fig. S6). The 0.6-kb GLI2-binding portion of Fragment 1a significantly increased transcription from the FOXF1 promoter in our reporter assay, further indicating that it may regulate FOXF1 in vivo (Fig. 3A,B). The increase of FOXF1 transcription occurred not only in cis but also in trans, suggesting that this activation could occur in vivo through chromatin looping that juxtaposes the FOXF1 promoter and SDR. The CTCF binding within the SDR (Supplemental Fig. S8) may contribute to the formation of this looping architecture (Phillips and Corces 2009). Both cis and trans activations of the FOXF1 promoter seem to be specific, since (1) substitution of the 0.6-kb sequence from the 1.5-kb Fragment 1a with a 1.4-kb fragment of Segment 2, harboring sites for transcription factors other than GLI2, in trans or in cis in its original genomic orientation had no effect on the promoter activity; and (2) the 3-kb empty vector pGEM-T Easy did not activate the FOXF1 promoter.
We supported the chromatin looping hypothesis via 4C studies, in which we identified physical interaction of the SDR with the FOXF1 promoter close to the Pol II–binding site (Fig. 3C). This interaction brings the distant GLI2-binding sites to the proximity of the FOXF1 promoter. Although only one of the distant GLI-binding sites of SDR perfectly matches the consensus “core” sequence, the clustering of nine of these sites within this segment may have synergistic effects on GLI2 binding. Moreover, the variant GLI-binding sites with relatively low affinity have been recently shown to strongly induce transcription when present in native promoters (Winklmayr et al. 2010). Since we did not detect interactions between SDR and the FOXF1 promoter in lymphoblasts, where FOXF1 is not expressed, we propose that chromatin looping between SDR and FOXF1 allows GLI2 to increase FOXF1 activity specifically in lung endothelium.
Importantly, we have also identified putative lncRNA genes within the SDR region (Fig. 2A). LncRNAs play a crucial role in embryonic development (Ulitsky et al. 2011). Moreover, lncRNAs have been proposed to play a role in imprinting and both in cis and in trans regulation of gene expression by acting as scaffolds for chromatin-modifying complexes and nuclear bodies, as enhancers as well as mediators of long-range chromatin interactions (Guttman et al. 2009; Mattick 2009, 2010; Nagano and Fraser 2011; Wang and Chang 2011). Thus, it seems tempting to speculate that some of the lung-expressed lncRNAs may be needed for tissue-specific interaction of the FOXF1 promoter with the distant regulatory regions. LncRNAs can also guide chromatin-modifying complexes to specific genomic loci to regulate gene expression (Mattick 2010). For instance, Khalil et al. (2009) showed that a 34-kb lncRNA FOXF1-AS1, mapping 1.7 kb upstream of FOXF1, binds the Polycomb repressive complex PRC2. Of note, this lncRNA has the highest expression in human lungs (http://genome.ucsc.edu) (see also Supplemental Fig. S1) and partially overlaps with the FOXF1 promoter, and its locus is largely deleted in the ACD/MPV case 41.4. Our preliminary experiments on FOXF1-AS1 knockdown using siRNAs showed that ∼50% decrease of FOXF1-AS1 expression in HPMEC resulted in a weak increase of FOXF1 transcription (Supplemental Fig. S1), supporting its suggested function as a transcriptional repressor (Khalil et al. 2009). However, in case 41.4, in addition to the FOXF1-AS1 gene, a GLI-binding consensus sequence is also deleted, likely leading to a net decrease of FOXF1 expression.
Intriguingly, all of the deletions for which we were able to determine the parental chromosome origin arose on maternal chromosome 16, strongly suggesting that FOXF1 could be paternally imprinted (P < 0.001, n = 14). The paternal imprinting of FOXF1 has been also predicted bioinformatically (Luedi et al. 2007). Even so, any paternal imprinting is probably incomplete, because paternal uniparental disomy of chromosome 16 (UPD16pat) has been reported in a child with only prenatal and postnatal growth retardation (Kohlhase et al. 2000). Our data showing differential expression of FOXF1 in the lungs support a partial imprinting hypothesis (Supplemental Fig. S10). We also reported a paternally inherited no-stop mutation (last codon) in FOXF1 in a patient with ACD/MPV (Stankiewicz et al. 2009). We hypothesize that the extended FOXF1 RNA transcript might have escaped nonsense-mediated decay and negatively interacted with the wild-type copy, resulting in ACD/MPV.
We have investigated in detail CpG methylation as a potential mechanism of FOXF1 imprinting. The FOXF1 promoter resides within a large CpG island (Lo et al. 2010), which is not methylated in normal lung tissue (Fig. 4A). Using genome-wide differential methylation array hybridization and bisulfate sequencing, we found that a small CpG island, located within Fragment 1a of SDR and overlapping with GLI-binding sites, is differentially methylated. Using in vitro methylation and reporter assays, we showed that the methylation of this distant CpG island compromises the ability of its GLI-binding region to activate the FOXF1 promoter in vitro (Fig. 4B). Although only two of the GLI-binding site variants include CpGs within their sequence and thus can be directly affected by CpG methylation, it is likely that methylation of the CpG island changes the local DNA environment, resulting in the decrease of affinity of its GLI-binding sites for GLI2. Interestingly, methylation of CpGs has been also shown to inhibit interaction with DNA of the GLI-type zinc-finger factor YYI (Kim et al. 2003).
Position effect or cis-ruption disorders are defined as conditions arising due to disruption of the cis regulatory genomic architecture of a disease gene locus (Kleinjan and van Heyningen 2005; Kleinjan and Coutinho 2009). To date, this phenomenon has been reported for more than 40 genetic loci in the human genome; however, the underlying mechanism remains unknown. Of interest, a 7.4-kb cis-regulatory deletion disrupting conserved noncoding sequences and their interaction with the promoter of another FOX gene, FOXL2, mapping more than 280 kb apart, has been described as pathogenic for blepharophimosis, ptosis, and epicanthus inversus (BPES, OMIM 110100) (D'haene et al. 2009). In addition, genomic deletions and an apparently balanced translocation breakpoint mapping over 250-kb downstream from FOXG1 have been reported in patients with a congenital variant Rett syndrome (OMIM 613454) (Kortüm et al. 2011; Ellaway et al. 2012), suggesting that also other FOX genes may be regulated by long-range chromatin cis-interactions. Recently, Guttman et al. (2011) successfully knocked down genome-wide 147 (out of 226) lncRNAs using shRNAs and observed gene dysregulation both in trans and cis, with cis effects ranging up to 300 kb. Our findings further suggest that lncRNA-controlled gene regulation could be responsible for the position effect phenomenon in humans and potentially cause many other disorders of development.
In summary, we propose a model of long-range regulation of FOXF1 expression in the lungs that includes CpG methylation-controlled GLI2 binding at a distant tissue-specific enhancer, whereby chromosomal looping, likely mediated by lncRNAs and/or CTCF, juxtaposes this enhancer and the FOXF1 promoter.
Methods
Subject recruitment
DNA samples were collected from nine unrelated probands clinically and histopathologically diagnosed with ACD/MPV. All patients died from severe pulmonary insufficiency and hypertension in the first month of life.
DNA and RNA isolation, and DNA sequencing
Peripheral blood DNA was extracted using the Gentra Puregene Blood Kit (QIAGEN). DNA from frozen lung tissues was extracted using the DNeasy Blood & Tissue Kit (QIAGEN). DNA from FFPE lung tissues was isolated following the manufacturer's protocol (Agilent Technologies). RNA from cultured HPMEC was extracted using the RNeasy Protect Mini Kit (Invitrogen). Lung RNA was isolated from frozen tissues using the Illustra TriplePrep Kit (GE Healthcare).
PCR products and plasmid DNA were sequenced by the Sanger method (Lone Star Labs). Before cycle sequencing, PCR products were treated with ExoSAP-IT (USB) to remove unincorporated nucleotides and primers.
DNA sequence analysis
Reference sequences were downloaded from the UCSC Genome Browser (NCBI build 37/hg19; http://genome.ucsc.edu). DNA sequence similarities were analyzed using BLAT (http://genome.ucsc.edu). Evolutionary conservation and regulatory potential of the analyzed sequences were assessed using the UCSC “Conservation” and “7× Reg potential” tracks, respectively. The 7× regulatory potential scores are computed from alignments of human, chimpanzee, macaque, mouse, rat, dog, and cow genomes using log-ratios of transition probabilities from two variable order Markov models (Kolbe et al. 2004; King et al. 2005). Sequences were assembled using Sequencher v4.8 (GeneCodes). Repetitive sequences were identified using RepeatMasker (http://repeatmasker.org). GC content was determined using CpGPlot (http://ebi.ac.uk/Tools/emboss/cpgplot). CTCF-binding hotspots were identified using the University of Washington CTCF binding site track from the UCSC Genome Browser (ChIP-Seq determined) using a threshold value of 500. P-values of finding microdeletion with both breakpoints in Alu family repetitive elements by chance were calculated using an approach similar to the one presented by de Smith et al. (2008) (see also Supplemental Methods).
Screening FOXF1 for mutations
The entire coding region of FOXF1 (two exons) and its splicing sites were amplified for each patient in partially overlapping fragments using GoTaq Flexi DNA Polymerase (Promega) in the presence of 8% DMSO. The PCR conditions were 25 cycles of incubation for 30 sec at 94°C, 30 sec at 56°C, and 30 sec at 72°C. Following purification, PCR products were directly sequenced. DNA sequences were then compared with the FOXF1 reference sequence (NM_001451.2).
Genomic copy-number analysis
CNVs were identified by aCGH with either custom-designed 16q24.1 region-specific (1 Mb flanking FOXF1) 180K oligonucleotide microarrays (Agilent Technologies) or 4.2M genome-wide oligonucleotide microarrays (Roche-NimbleGen). 4×180 microarrays were scanned using an Agilent DNA Microarray Scanner, and the data were analyzed using Agilent Genomic Workbench software. 4.2M microarrays were scanned on a Roche-NimbleGen MS 200 Microarray Scanner. Scanned images of the arrays were processed using NimbleScan v2.5 (Roche NimbleGen) and analyzed using SignalMap v1.9 (Roche NimbleGen).
Amplification and sequencing of microdeletion breakpoints
PCR primers flanking each microdeletion (Supplemental Table S4) were designed using Primer3 v0.4.0 software (http://frodo.wi.mit.edu/primer3/). Amplification of 5- to 12-kb junction fragments for sequencing was performed using LA Taq polymerase (TaKaRa Bio USA).
Parental origin of microdeletions
Parental origin of microdeletions was determined following identification of informative SNPs in parental and patients' chromosomes. Regions containing the potentially informative SNPs were amplified by PCR and directly sequenced. The primers used in this analysis were designed based on the location of known SNPs.
ChIP-chip analysis
Chromatin immunoprecipitation with anti-GLI2 antibody was conducted in HPMEC (ScienCell Research Laboratories). Expression of FOXF1 and GLI2 in these cells was confirmed by RT-PCR using the SuperScript One-Step RT-PCR Kit (Invitrogen) (Supplemental Fig. S7; Supplemental Table S5). The reference DNA was extracted from skin fibroblasts that do not express FOXF1. HPMEC were cultured to confluence in EMS medium supplemented with 10% FBS, ECGS, and 1% penicillin-streptomycin (ScienCell Research Laboratories) at 37°C with 5% CO2. Fibroblasts were cultured in DMEM medium (Invitrogen), supplemented with 10% FBS and 1% penicillin–streptomycin, at 37°C with 10% CO2. Because of low levels of endogenous GLI2 expression, cells were transfected, 24 h before the assay, with GLI2-expressing plasmid pCS2Gli2 (Roessler et al. 2005) (Addgene) (1 μg of DNA/60-mm plate) using Lipofectamine LTX and Plus Reagent (Invitrogen). Protein–chromatin interactions were captured by incubating cells with 1% formaldehyde for 10 min at room temperature. Cells were then lysed in SDS lysis buffer and sonicated. Fragmentation of the DNA to ∼1-kb fragments was verified by agarose gel electrophoresis. Immunoprecipitation was performed using an EZ ChIP kit (Millipore) and anti-GLI2 antibody (R&D Systems). Five microliters of the anti-GLI-2 antibody was added to 1 mL of the sample, and the GLI2–DNA complex was immunoprecipitated overnight at 4°C. Twenty microliters of Normal Sheep IgG (R&D Systems) was used as a negative control. Anti-mono/di/tri methyl-histone H3 (lys4) clone AW304 rabbit monoclonal IgG (Upstate) was used as a positive control. The MinElute PCR Purification Kit (QIAGEN) was used to purify and concentrate DNA to a final volume of 10 μL. Immunoprecipitated DNA was then amplified using the GenomePlex Complete Whole Genome Amplification Kit (Sigma-Aldrich). After amplification, samples were labeled with Cyanine dUTP (Agilent Technologies) using the Agilent Genomic DNA Enzymatic Labeling Kit and used for array hybridization for 24 h at 65°C on a custom-designed 16q24.1-region-specific 4x180K oligonucleotide microarray (Agilent Technologies).
Reporter assay constructs
The −5.5-kb FOXF1 promoter region (chr16: 86,538,679–86,544,175), directly upstream of the FOXF1's AUG initiation codon, was amplified by PCR from genomic DNA isolated from the blood sample of a normal control individual. This region contained the entire 43-bp-long 5′-untranslated part of the FOXF1 first exon. The primers used for the amplification were Foxp2F2: 5′-CTAGCTAGCACATTTCCTCATATTCTGTGTAGAGAGCACCT-3′ and FoxAUG2R: 5′-TTGCGCCGATTCGAACGGGTGGCTGCTG-3′, and included restrictions sites for NheI and BstBI, respectively. PCR was done using LA Taq DNA polymerase (TaKaRa Bio) in the presence of 6% DMSO, applying 25 cycles of 30 sec at 94°C and 5 min at 68°C. Following digestion with NheI and BstBI (NEB), the PCR product was cloned between NheI and BstBI sites of the multiple cloning site of the promoterless SEAP reporter vector, pSEAP-Basic (Clontech), generating a plasmid, p5.5FoxSEAP. The pSEAP2-Control vector, containing the constitutive SV40 early promoter with its enhancer, was used as a positive control in the assay. The plasmid p41.4Δ4, containing the truncated version of the 5.5-kb FOXF1 promoter region (devoid of its 5′ portion deleted in an ACD/MPV case 41.4) was constructed by digesting the p5.5FoxSEAP with KpnI and relegating the remaining portion of the vector. A 0.6-kb portion of Fragment 1a (chr16:86,232,261–86,232,908), bearing CpG island and GLI2-binding sites, was amplified from genomic DNA of a normal control individual using Taq DNA polymerase (Invitrogen) and the following primers: upsdelCGliF: 5′-GTGCTAGCATGGTGTAAATCACCCTGACAGTAAACACT-3′ and upsdelCGliR1: 5′-GTGCTAGCGATCTATTCTGCTATCTACCCTCTGCTTTTCC-3′, both encompassing the NheI site. PCR conditions were 25 cycles of 30 sec at 94°C, 30 sec at 58°C, and 1 min at 72°C. This 0.6-kb fragment was then cloned both into pGEM-T Easy (Promega) by T-vector cloning to create pGliTEasy, and, following digestion with NheI, into the NheI site of p5.5FoxSEAP, upstream of the 5.5-kb FOXF1 promoter region, generating pGli-5.5FoxSEAP. The 1.4-kb putative enhancer region of Segment 2 (chr16:86,256,619–86,258,038) was amplified by PCR using Pfu Ultra DNA polymerase (Stratagene) with the primers upsdelEnhF: 5′-AAACTTCATCTCTCCCTGCAGCTTCTCTGT-3′ and upsdelEnhR: 5′-GTTGGACTCTGAAATCAGTGCCTTCAACAT-3′. The blunt-ended PCR product was A-tailed using GoTaq Flexi DNA polymerase and cloned in the pGEM-T Easy vector to generate plasmid pEnhTEasy. This new vector was then cut with NheI, and the putative enhancer-containing fragment was subcloned in the NheI site of p5.5FoxSEAP, generating the plasmid pEnh5.5FoxSEAP.
Cell transfection and reporter assay
The HPMEC were cultured as described above. Transient transfection of HPMEC with reporter gene constructs was done by electroporation using the NEON transfection system (Invitrogen). The electroporation parameters were as follows: pulse voltage, 1350 V; pulse width, 30 msec; pulse number, 1; cell density, 1.0 × 107 cells/mL. The transfection efficiency was between 20% and 30%. One-half microgram of pSEAP2Basic (no promoter), pSEAP2Control (Clontech), pEnhTEasy, pUpstdelGliTEasy, or 1 μg of p5.5FoxSEAP, pGli5.5FoxSEAP, or pEnh5.5FoxSEAP was cotransfected with 0.1 μg of pMetLuc-Control (Clontech) (internal control for transfection efficiency) and with or without 0.4 μg of pCS2Gli2 in 24-well plate setting.
In an in vitro methylation experiment, 3 μg of linearized, methylated, or nonmethylated pUpstdelGliTEasy vector was cotransfected with p5.5FoxSEAP and pMetLucControl. One set of transfections included also pCS2Gli2. Each well of 24-well plates was seeded with 1.5 × 105 cells. Activities of SEAP and Metridia luciferase (MetLuc) were measured 55 h later by a luminescence assay using the Ready-to-Glow Dual Secreted Reporter System (Clontech) according to the manufacturer's protocol. Luminescence was recorded using microplate LUMIstar Omega luminometer (BMG Labtech). SEAP activities of individual transfections were normalized for Metridia luciferase activities. Results are shown as the mean of three independent experiments. Statistical significance of the observed differences was estimated applying an unpaired t-test.
4C analysis
3C libraries were generated according to the protocol described by Dostie and Dekker (2007) using 1 × 107 HPMEC or lymphoblasts. Cross-linked DNA was digested with EcoRI overnight, and religated with T4 ligase for 4 h at 16°C at low DNA concentration. The 3C library was then processed according to the procedure described by Simonis et al. (2007). The ligation junctions were trimmed by digestion of 3C library with TaqaI overnight, followed by phenol:chloroform extraction, religation at low DNA concentration for 4 h at 16°C, and DNA linearization with HindIII overnight at 37°C. The 4C template was created by linear amplification of DNA using the Expand Long Template PCR System (Roche Applied Science). Two hundred nanograms of 4C template was amplified per reaction. The primers used for amplification were 5′-GGCAGGAAGTTTACAGGGTTTAACG-3′ and 5′-TGTGTGTGCTAATGTGTGGACAAGA-3′. The primers were designed within the EcoRI-TaqaI fragment (chr16:86,542,571–86,543,312), containing part of the Pol II binding site. The PCR cycling conditions were 30 sec at 94°C, 1 min at 55°C, and 3 min at 68°C. Sixteen reactions were conducted for each template. Pooled PCR products were purified using the MinElute PCR Purification Kit (QIAGEN).
As a reference DNA for hybridization, we used uncross-linked DNA isolated from HPMEC or lymphoblasts with Puregene Blood Core Kit A, and digested overnight with EcoRI and TaqaI.
Custom-designed 720K microarrays covering 2-Mb regions flanking FOXF1 were designed and produced by Roche-NimbleGen. Labeling and hybridization of 4C DNA libraries were done according to the manufacturer's instructions (Roche-NimbleGen). The arrays were scanned on a NimbleGen 200 Microarray Scanner, and the data were analyzed using NimbleScan v2.5 and SignalMap v1.9. The 4C peak data were generated from the scaled log2 ratio data. Peaks were detected by searching four or more probes whose signals are above the specified cutoff values, using a 500-bp sliding window. Each peak was assigned a false discovery rate (FDR) score. Scores ≤0.2 are indicative of an interaction site.
Bisulfite sequencing
Bisulfite modification of lung and blood DNA for sequencing was performed using the EpiTect Bisulfite Kit (QIAGEN). Primers for methylation PCR were designed using MethPrimer software (http://urogene.org/methprimer/index1.html) (Supplemental Table S6; Li and Dahiya 2002). PCR was performed in a 25-μL reaction mixture containing 200 ng of bisulfite-treated DNA, 0.5 nmol of each primer, and 1.25 units of HotStarTaq polymerase (QIAGEN). The cycling conditions were 1 min at 95°C, 30 sec at 50°C–62°C, and 1 min at 72°C. PCR products ranging from 0.2 to 0.6 kb were purified from unincorporated primers and nucleotides using the QIAquick PCR Purification Kit (QIAGEN), and T-vector cloned in DH5α cells using the pGEM-T Easy Vector System. Plasmid minipreps were prepared from 25 randomly selected transformant colonies using the Perfectprep Plasmid Mini Kit (Eppendorf) and sequenced using T7 and SP6 promoter universal primers.
In vitro methylation of CpG island
For the in vitro methylation assay, the pUpstdelGliTEasy vector was first linearized using the SphI restriction nuclease (NEB) and then treated with HpaII methyltransferase (NEB), which methylates the internal C residue of the sequence CCGG. The 25-μL reaction mixture contained 1 μg of the DNA, 1 unit of HpaII methylase, 50 mM Tris-HCl (pH 7.5), 10 mM EDTA, 5 mM 2-mercaptoethanol, and 80 μM S-adenosylmethinine. Methylation was performed for 1 h at 37°C. In the mock-methylation reaction, S-adenosylmethinine was omitted. Plasmid DNA was subsequently extracted with phenol:chlorophorm, precipitated with isopropanol, and analyzed for the degree of methylation by digestion with HpaII restriction nuclease (NEB). Completely methylated plasmid DNA was then used for transfections; unmethylated DNA was used as a control. The stability of methylation following transfection of the methylated construct was demonstrated recently in the similar experiments reported by Ilan and Katzav (2012) and Matousková et al. (2006).
Analysis of FOXF1 expression from individual parental chromosomes
To determine whether there is a difference in FOXF1 expression from maternal versus paternal chromosomes, we first looked for heterozygous SNPs in FOXF1 exons in genomic DNA extracted from normal newborn lung tissue, from which we also extracted RNA. We identified heterologous SNP G>G/A (chr16:86,547,496) within FOXF1 exon 2. We then performed two sets of RT-PCR, using RNA with reverse primer in one setting ending in a G nucleotide (FoxE2RnaRG: 5′-CAGAAAGTTTACAGTAGAGGTTGGG-3′) and, in the other setting, in an A nucleotide (FoxE2RnaRA: 5′-CAGAAAGTTTACAGTAGAGGTTGGA-3′). The forward primer was the same in both settings (FoxE2RnaF1: 5′-GTCTCCCTTTAGAGCCGTCTTTTG-3′). Both pairs of primers were checked for their equal efficiency in priming PCR using genomic DNA. Semiquantitative RT-PCR was done using the SuperScript One-Step RT-PCR Kit. cDNA was synthesized for 30 min at 50°C. The temperature profile of PCR was 19 cycles of 30 sec at 94°C, 45 sec at 56°C, and 45 sec at 72°C, and a final extension for 5 min at 72°C.
Data access
The aCGH, ChIP-chip, and 4C data sets can be accessed through the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE39258.
Acknowledgments
We thank C. Brown for the access to the electroporation system; M. Withers for technical assistance; C. Dugauquier for histopathological analysis; and J. Bondaruk, A. Gambin, T. Gambin, V.V. Kalinichenko, J.R. Lupski, A. Rodriguez, and C. Shaw-Smith for helpful discussion. Part of this work was supported by NIH grant 1RO1HL101975-01 to P. Stankiewicz, a NORD grant and Pilot Project Award from the Texas Children's Hospital to P. Sen, and the Polish National Science Center grant 2011/01/B/NZ2/00864 and the European Social Fund (UDA-POKL.04.01.01-00-072/09-00) to P.D.
Footnotes
-
↵23 Corresponding author
E-mail pawels{at}bcm.edu
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.141887.112.
- Received April 16, 2012.
- Accepted September 17, 2012.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








