
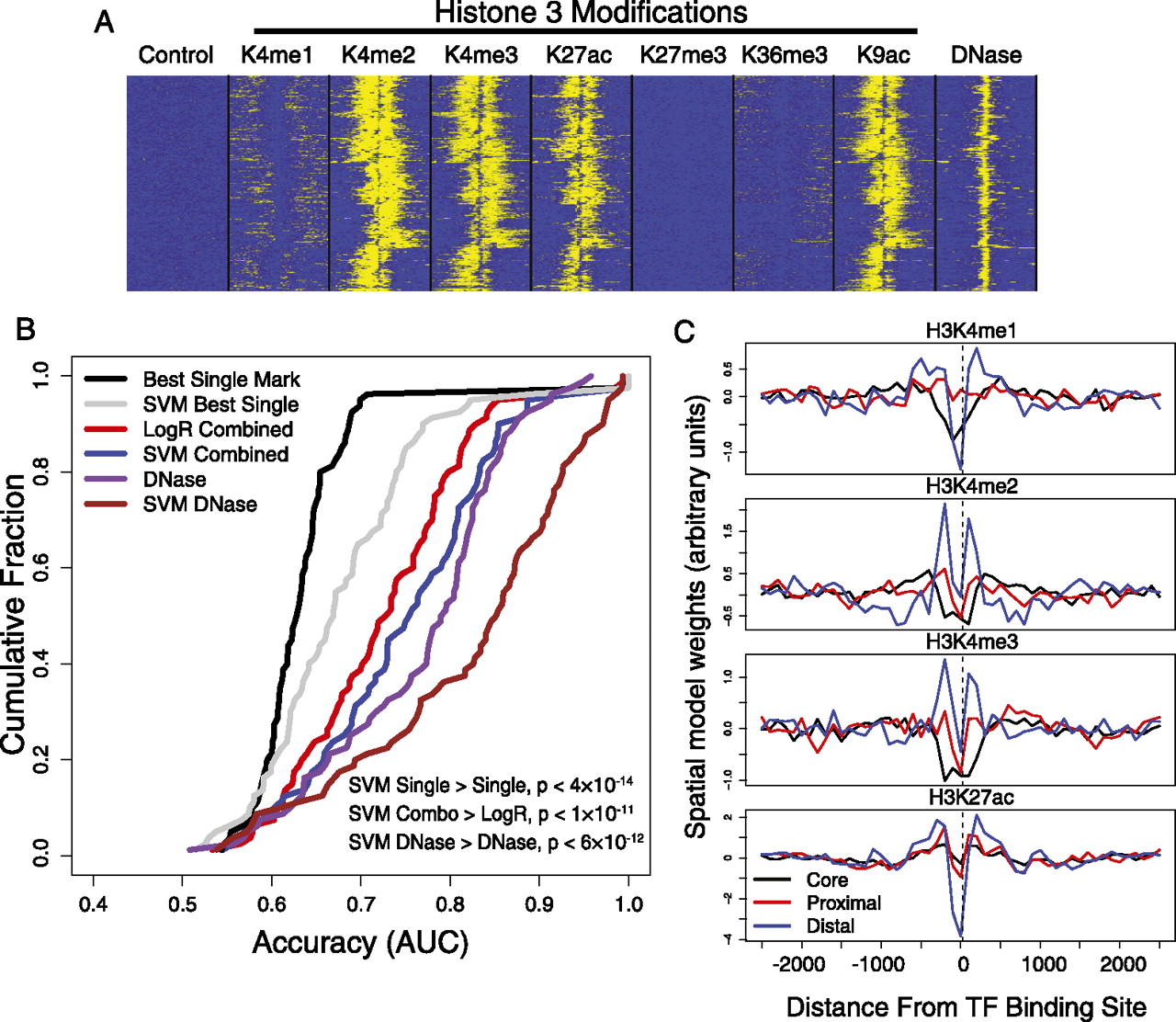
SVM spatial chromatin models better predict binding sites than simpler models. (A) The distribution of histone marks over 5000-bp windows centered at GABPA ChIP-seq peaks in K562 shows spatial organization of multiple correlated signals. (B) The accuracy of multiple chromatin models suggests that spatial signatures of DNase accessibility better predict binding sites than other methods. The cumulative distributions of prediction accuracy (AUC; x-axis) across a subset of ChIP-seq experiments are shown for multiple chromatin representations. Shown are an SVM model trained on all spatially binned histone marks (blue), which is more accurate than standard ranking based on best single mark read counts (black) or a logistic regression combination of read counts (red); similarly, an SVM model trained on spatially binned DNase-seq reads (brown) better describes binding sites than use of DNase bin counts (purple). Paired signed rank test P-values are shown. (C) Transcription factors that bind the core promoter, proximal to transcript start site, or distal to start site have distinctive spatial patterns of histone modifications. The four plots show spatial coordinates of the learned bin weights arranged along the x-axis, with the values of the weights shown on the y-axis. The bin weightings are averaged across subsets of core, proximal, and distal binding transcription factors. The valleys at the binding site suggest that spatial models are capturing predictive information regarding the differential spacing of nucleosome-depleted regions at core, proximal, and distal binding sites.








