
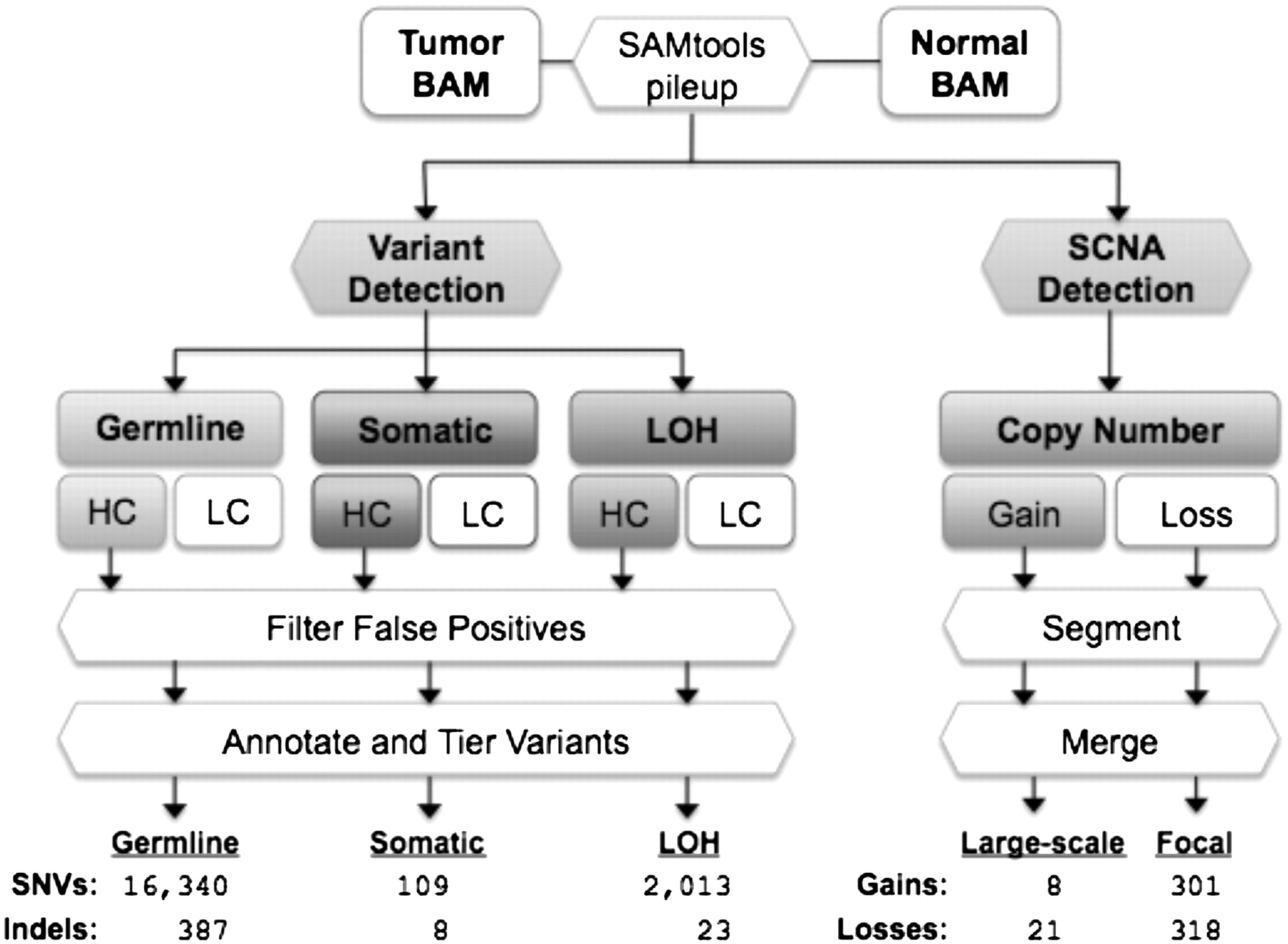
The VarScan 2 mutation and copy number alteration detection algorithms. Alignments in BAM format for a tumor–normal pair are read simultaneously to identify inherited (germline), loss-of-heterozygosity (LOH), and somatic mutation events. Variants in each category are further classified as high confidence (HC) or low confidence (LC). HC variants are filtered to remove false positives from common sequencing- and alignment-related artifacts (see Table 1). The resulting variants are annotated and organized by tier; the average number of “tier 1” coding variants per tumor is shown for each category. At positions with at least 20× coverage (default), copy number alterations are detected by comparison of Q20 read depths from matched tumor–normal pairs, normalized based on the amount of input data for each sample. Raw contiguous regions from VarScan 2 are processed by circular binary segmentation (CBS) and a subsequent merging procedure that joins adjacent segments yields a set of somatic copy number alterations, which are further classified as large-scale (>25% of chromosome arm) or focal (<25%) events. Shown are the average numbers of events detected in 142 ovarian exomes.








