
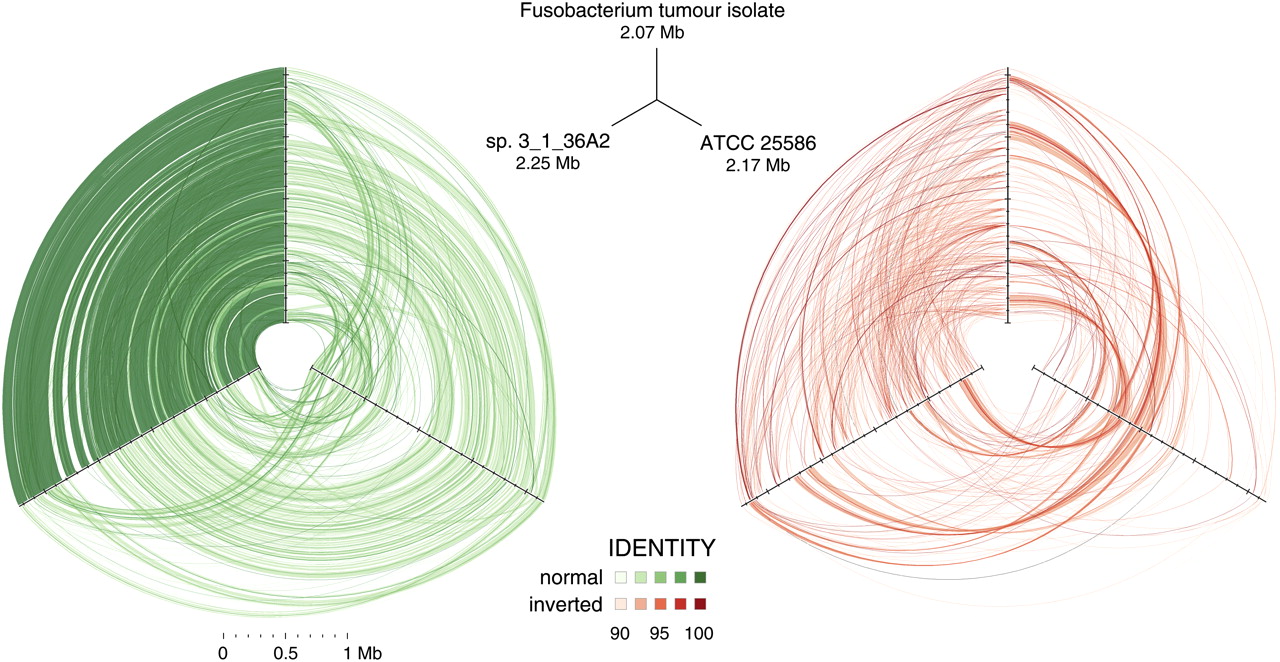
Hive plots showing alignment of three Fusobacterium genomes. Approximately 32 million high-quality WGS Illumina HiSeq reads (≥99 consecutive Q30 bases) from Fusobacterium tumor isolate CC53 were assembled with SSAKE (v3.7, default options) into 379 contigs. The contigs were aligned using cross_match (-minmatch 29 -minscore 59 -masklevel 101) to the complete F. nucleatum susb. nucleatum ATCC 25586 genome and, independently, to the 12-contig HMP Fusobacterium sp. 3_1_36A2 assembly, respectively; and ordered/oriented based on the highest identity to the latter sequence. Three-way cross_match (http://www.phrap.org) alignments between each Fusobacterium genome were performed and represented visually using hive plots (http://www.hiveplot.com). For each, the top, left, and right axes are proportional to genome size and represent the Fusobacterium tumor isolate CC53 (2.07 Mb), the HMP sp. 3_1_36A2 (2.25 Mb), and the ATCC 25586 type strain (2.17 Mb), in that order. Synteny between the isolates is depicted by green and red links that show direct and inverted alignments, respectively. Sequence similarity and synteny is highest between CC53 and sp. 3_1_36A2, as evidenced by a greater density of high similarity sequence matches between them, relative to ATCC 25586, and shared patterns of inversions compared to this reference strain. Three regions of sequences present in sp. 3_1_36A2 but absent from CC53 are apparent as conspicuous gaps on the sp. 3_1_36A2 axis. Sequence segments unique to CC53 are not visible at this scale.








