
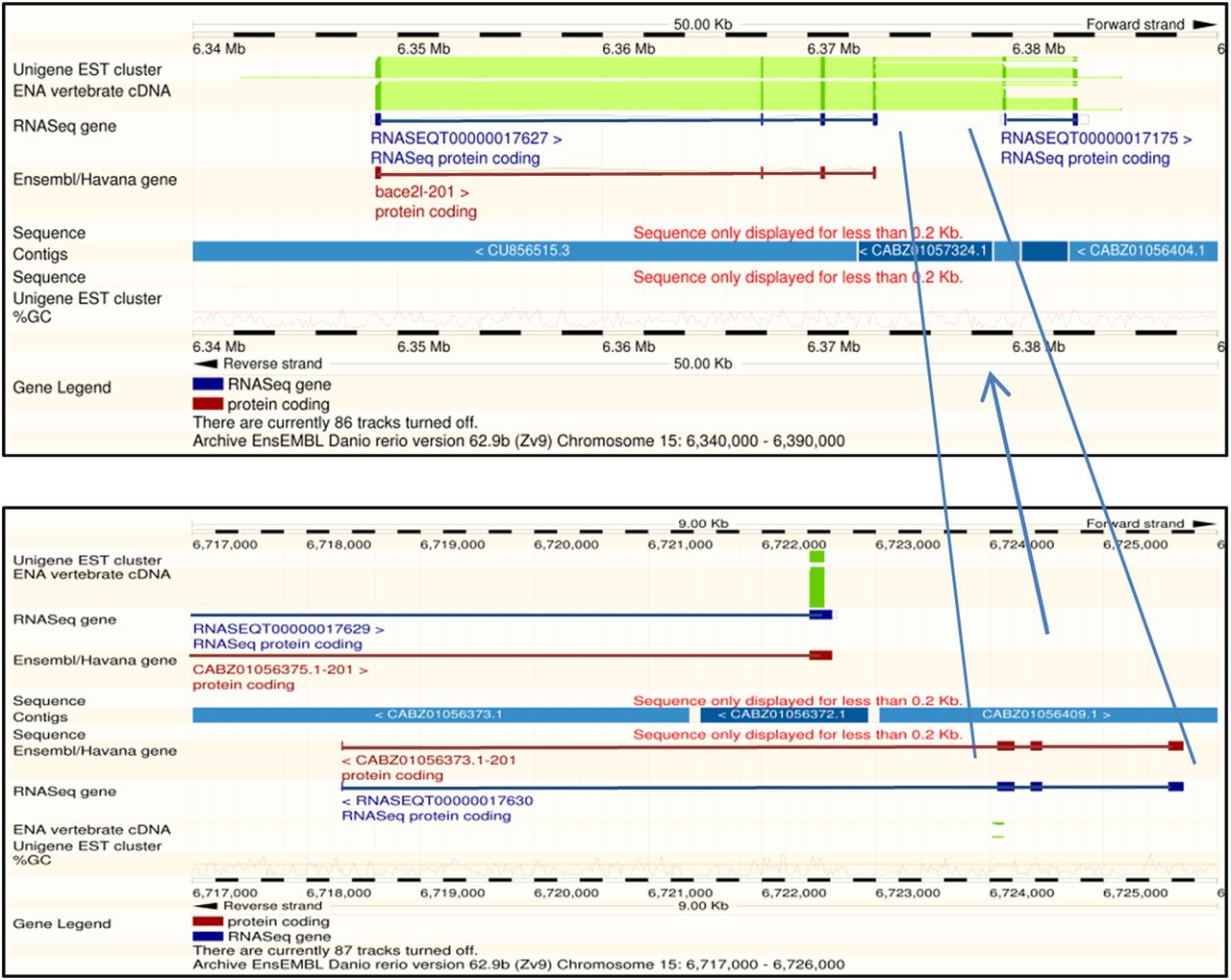
The Ensembl browsers from release 62 between 15: 6,348,671-6,373,413 shows the transcript ENSDART00000065824 for the gene bace2. This transcript was annotated from species-specific cDNAs BC164206.1, BC083415.1, and BC098874.1. Matching BC098874.1 to the Zv9 assembly using Ensembl suggests that the 5′ end matches four exons in the reference sequence as annotated in ENSDART00000065824, the central portion matches four exons over 340,000 bases away between 15:6,718,308 and 6,725,702, and the 3′ end matches two exons adjacent to the first four exons between 15:6,379,504 and 6,383,732 (note that the sequence of the short fourth exon of the central portion can be found at the 5′ end of the first exon of the final two exons and is probably an artifact of the read alignment process). This shuffled exon order suggests a reference sequence assembly problem. On closer inspection, apart from the first three exons, the transcript is aligned to capillary and Illumina whole-genome shotgun assemble rather than the more reliable genomic clone sequence. The underlining reference sequence of the central four exons appears to be incorrectly located on chromosome 15. The Ensembl genebuild has create the longest single transcript from the cDNA. Interestingly, the RNA-seq-only genebuild has a model in all three locations. The first four exons match RNASEQT00000017627, the middle three exons match RNASEQT00000017630 creating a partial additional gene, and the final two exons match RNASEQT00000017175. The RNA-seq genebuild is unable to join the first four exons to the last two exons because three exons and four introns are missing. This example demonstrates annotation problems associated with errors in the reference sequence. Expanding this principle to other species, the degree of disrupted transcripts will be related to the quality of the assembly. It also highlights how a break in transcript contiguity can be caused by incomplete exon or intron data that could arise, for example, from low read coverage.








