
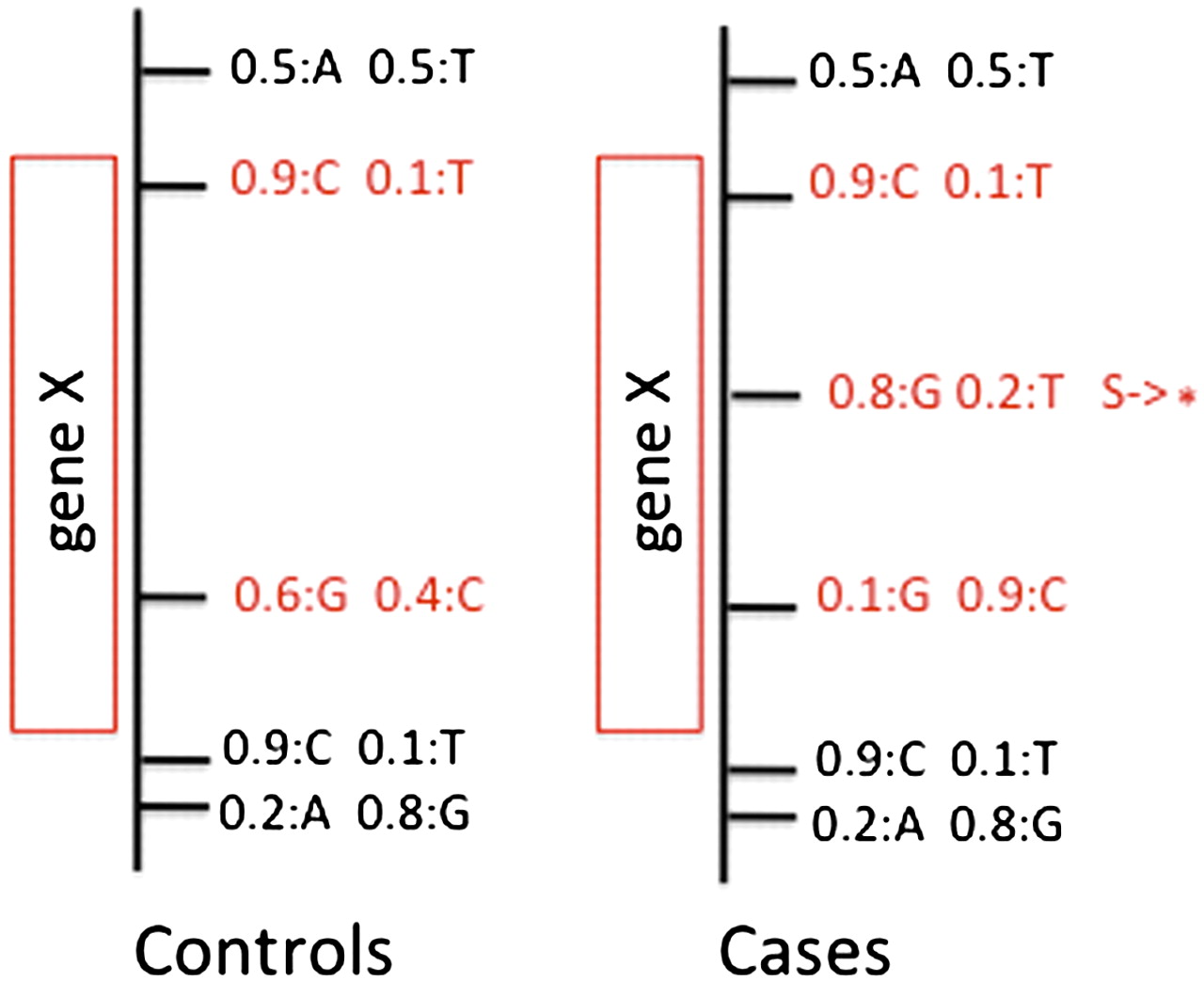
VAAST uses a feature-based approach to prioritization. Variants along with frequency information, e.g., 0.5:A 0.5:T, are grouped into user-defined features (red boxes). These features can be genes, sliding windows, conserved sequence regions, etc. Variants within the bounds of a given feature (shown in red) are then scored to give a composite likelihood for the observed genotypes at that feature under a healthy and disease model by comparing variant frequencies in the cases (target) compared to control (background) genomes. Variants producing nonsynonymous amino acid changes are simultaneously scored under a healthy and disease model.








