
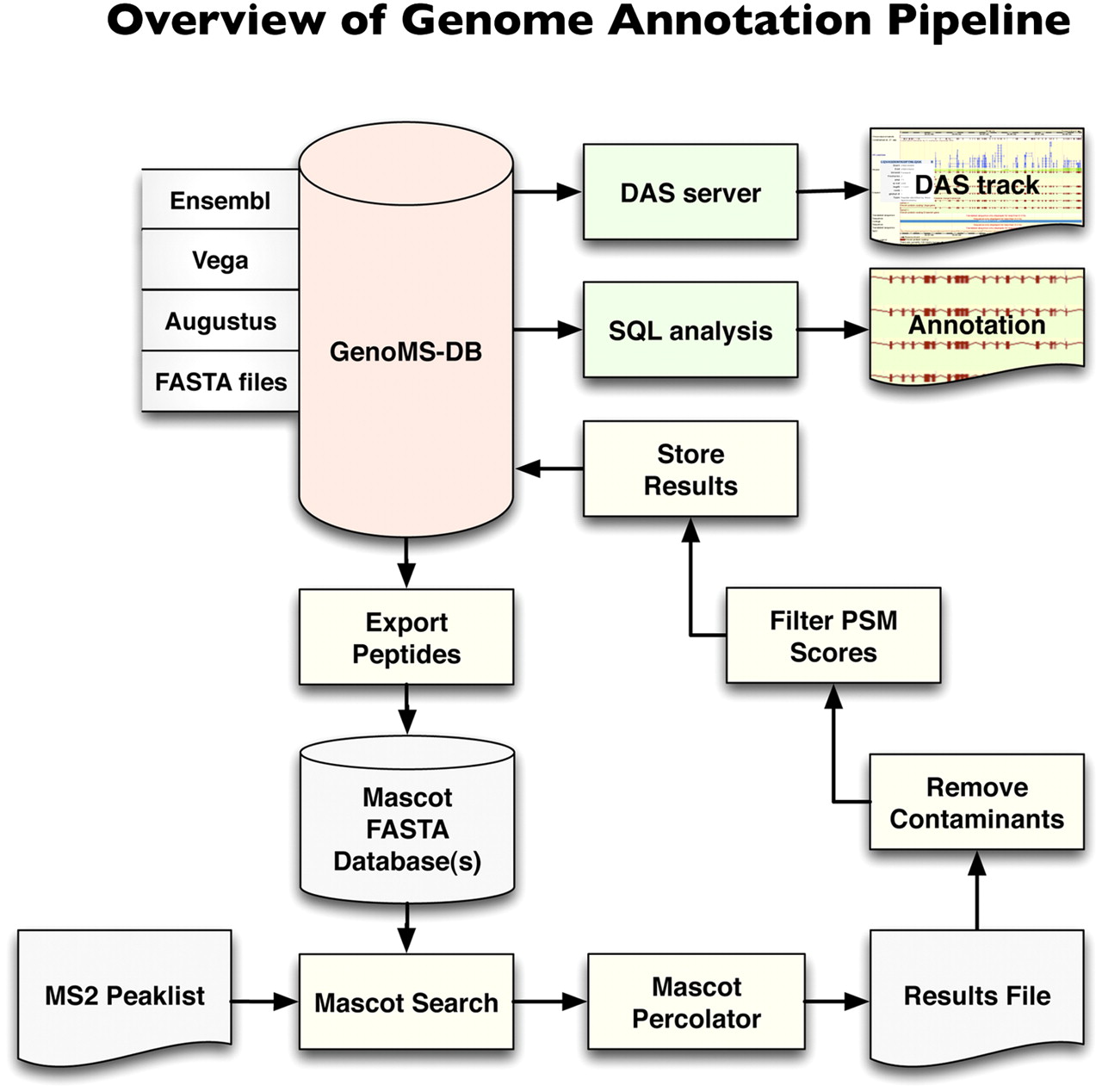
Genome annotation pipeline. The database at the core of the system, GenoMS-DB, is built by integrating all peptides that are derived from an in silico digestion of available data sources (Ensembl, Vega, Augustus). Each peptide derived from these data sources is associated with its genomic locus and context (such as gene, transcript, exon, or splice site information). Peptides from FASTA protein databases can optionally be integrated but would lack genome mapping. A set of nonredundant in silico digested peptides is exported from GenoMS-DB to create the Mascot search database. Tandem MS spectra are searched with Mascot and post-processed with Mascot Percolator to derive accurate probabilities on a per PSM basis. A series of steps removes common contaminant sequences and low-scoring PSMs from the results, prior to storing the remaining identifications into the GenoMS-DB database. This integration of peptide-genome mapping together with peptide identifications enables streamlined analysis with standard SQL or visualization as a track in a genome browser via a DAS feature server. This is a flexible pipeline where alternative gene prediction tools could be used to provide source peptides, and alternative search engines and probability assessment algorithms could be integrated.








