
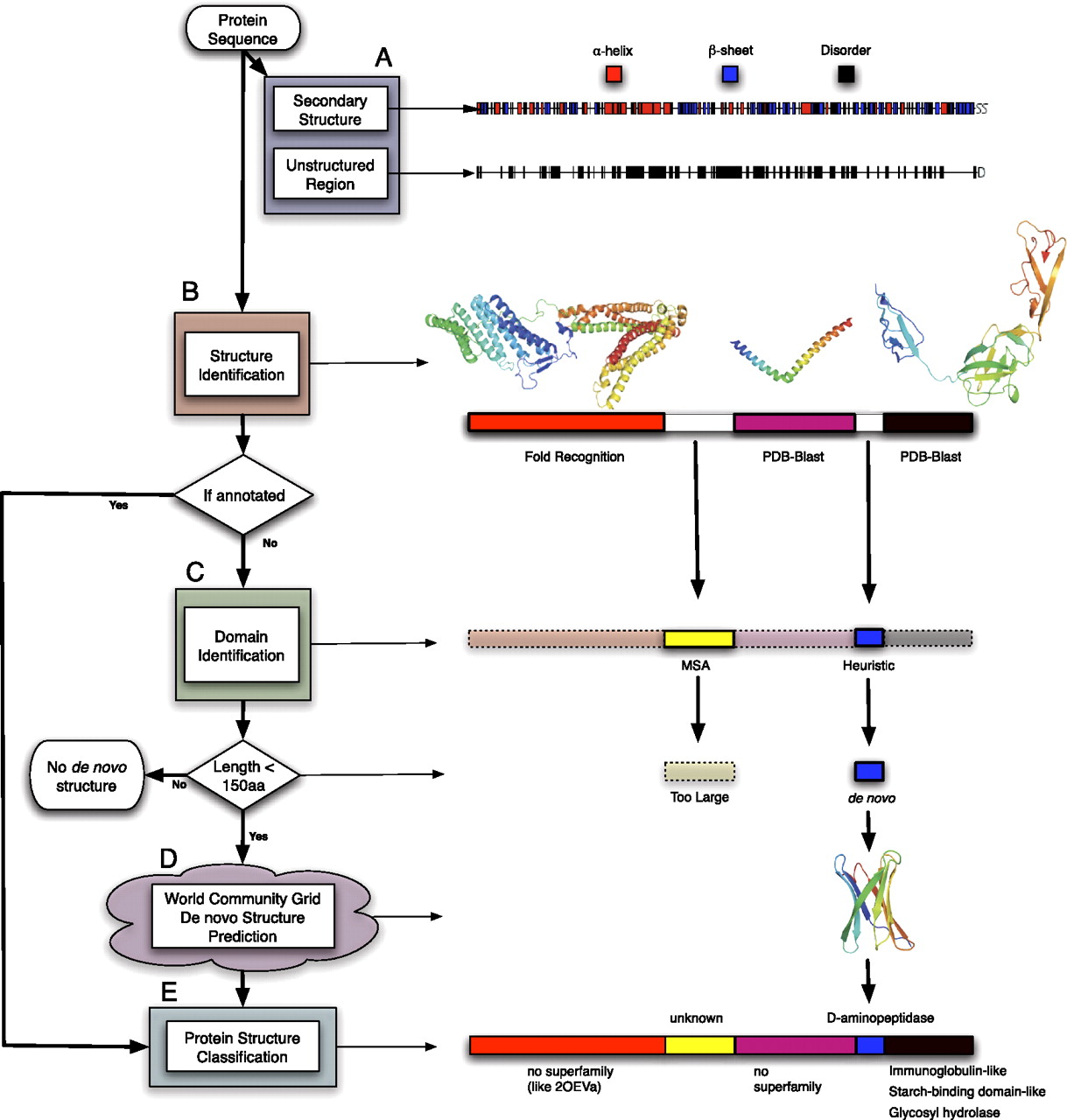
Flow diagram of the proteome folding pipeline using Lactobacillus prophage hypothetical protein Ljo_0324 as an example. (A) The Ljo_0324 sequence is first annotated with primary and secondary structure (PSIPRED, DISOPRED, and [not shown] TMHMM, COILS, and SignalP). (B) PDB-BLAST (PSI-BLAST) and fold recognition (FFAS03) are then used to compare the input sequence to the PDB database to identify a similar sequence/structure. (C) Regions of the protein sequence still unannotated are processed by Pfam, multiple sequence alignments (MSA), and a heuristic method for domain identification. (D) Domains <150 amino acids are then sent to the computational grid, where the three-dimensional structure is predicted by Rosetta. (E) Finally, domains with structural annotations (de novo, BLAST, or fold recognition) are classified into SCOP superfamilies. Regions annotated in each level of the figure are outlined in black, and regions annotated in previous levels are outlined in dotted lines. This example can be found online at http://www.yeastrc.org/pdr/viewProtein.do?id=2155068.








