
Abstract
Second-generation sequencing platforms have revolutionized the field of ancient DNA, opening access to complete genomes of past individuals and extinct species. However, these platforms are dependent on library construction and amplification steps that may result in sequences that do not reflect the original DNA template composition. This is particularly true for ancient DNA, where templates have undergone extensive damage post-mortem. Here, we report the results of the first “true single molecule sequencing” of ancient DNA. We generated 115.9 Mb and 76.9 Mb of DNA sequences from a permafrost-preserved Pleistocene horse bone using the Helicos HeliScope and Illumina GAIIx platforms, respectively. We find that the percentage of endogenous DNA sequences derived from the horse is higher among the Helicos data than Illumina data. This result indicates that the molecular biology tools used to generate sequencing libraries of ancient DNA molecules, as required for second-generation sequencing, introduce biases into the data that reduce the efficiency of the sequencing process and limit our ability to fully explore the molecular complexity of ancient DNA extracts. We demonstrate that simple modifications to the standard Helicos DNA template preparation protocol further increase the proportion of horse DNA for this sample by threefold. Comparison of Helicos-specific biases and sequence errors in modern DNA with those in ancient DNA also reveals extensive cytosine deamination damage at the 3′ ends of ancient templates, indicating the presence of 3′-sequence overhangs. Our results suggest that paleogenomes could be sequenced in an unprecedented manner by combining current second- and third-generation sequencing approaches.
Ancient DNA (aDNA) research began in the mid-eighties, when short mitochondrial DNA (mtDNA) fragments were successfully cloned and sequenced from museum specimens of the quagga (Equus quagga)—an equid that became extinct in South Africa at the end of the 19th century. The findings demonstrated that trace nucleic acids survive at least over the time frame of human history (Higuchi et al. 1984). The advent of the polymerase chain reaction (PCR) (Saiki et al. 1985), which allowed the retrieval of even single surviving molecules (Paabo et al. 1989), together with the finding of aDNA molecules preserved in both soft tissues and calcified material such as bones and teeth (Hagelberg et al. 1989), further advanced the field. Over the past two decades, aDNA has been shown to survive for at least a half-million years under frozen conditions (Willerslev et al. 2004; Johnson et al. 2007) and has been applied successfully to a range of biological questions, including reconstructing past animal population dynamics (e.g., Shapiro et al. 2004; de Bruyn et al. 2009; Campos et al. 2010, Stiller et al. 2010), paleoecosystems (e.g., Kuch et al. 2002; Willerslev et al. 2003, 2007), and prehistoric human migrations (e.g., Gilbert et al. 2008; Bramanti et al. 2009; Malmstrom et al. 2009; Haak et al. 2010), to infer past phenotypic traits and evolutionary relationships (e.g., Rohland et al. 2007, 2010), and even to re-examine the extinction date of megafaunal species (Haile et al. 2009).
The survival of aDNA in organic material is limited ultimately by processes of chemical damage that take place post-mortem. These commonly include hydrolytic and oxidative processes that fragment the DNA molecules into short pieces often not longer than 50–150 bp and change the biochemical structure of both the nucleotide bases and sugar-phosphate backbone (Paabo 1989; Hoss et al. 1996). As a result, damage-free modern DNA molecules can easily outcompete homologous ancient fragments during PCR, making aDNA studies highly prone to contamination (Paabo et al. 2004; Gilbert et al. 2005; Willerslev and Cooper 2005). Additionally, nucleotides are misincorporated by DNA polymerases while amplifying damaged templates, particularly at sites where cytosine has been deaminated to uracil, as the latter is the chemical analog of thymine, resulting in artefactual G/C to A/T mutations (so-called type II damage) (Paabo et al. 1989; Hoss et al. 1996; Hansen et al. 2001; Hofreiter et al. 2001; Gilbert et al. 2003, 2007a; Binladen et al. 2006; Stiller et al. 2006). Cytosine deamination is therefore a common feature of all aDNA templates, with misincorporation rates that can exceed real biological mutation rates and generate spurious sequence results (Ho et al. 2007). In early studies the need for cloning/sequencing of amplicons to filter out damage (Hofreiter et al. 2001) coupled with “requirements” of sequence replication in independent laboratories (Cooper and Poinar 2000) made the study of large numbers of samples financially prohibitive.
Since many mtDNA genomes coexist within each cell, any single mtDNA locus is represented by a much higher number of templates than are nuclear loci (Poinar et al. 2003). Therefore, the majority of aDNA research to date has focused on the recovery and analysis of short mtDNA fragments in order to maximize the chances of recovery. However, the information gained from mtDNA can be limited both by its maternal inheritance and its relatively high mutation rate compared with nuclear DNA.
In the last decade, a series of innovative methods have been developed in order to improve analysis of aDNA molecules. One of the first examples was a two-round multiplex PCR approach that substantially increased the amount of aDNA recovered from extracts; this approach was used to sequence complete mitogenomes and nuclear genes from Pleistocene-aged samples, improving phylogenetic inference and molecular estimates of species divergence (Krause et al. 2006) and providing phenotypic information such as skin color (Rompler et al. 2006). A second example was single primer extension (SPEX), a tool that has provided access to any preserved fragment at a given locus regardless of its length and, therefore, considerably improved genotyping accuracy (Brotherton et al. 2007).
However, no single methodological development has had the enormity of impact as the recent advent of second-generation sequencing technologies. These promoted a new era in the field of aDNA by opening access to complete mtDNA and nuDNA genomes from past individuals (Rasmussen et al. 2010) and extinct species (Green et al. 2006, 2008, 2010; Poinar et al. 2006; Gilbert et al. 2007b, 2008; Miller et al. 2008; Krause et al. 2010a; Reich et al. 2010). Massively parallel sequencing platforms such as 454 Life Sciences (Roche) GS FLX and Illumina GAIIx outcompete Sanger-based sequencing by several orders of magnitude (Green et al. 2006; Noonan et al. 2006). These sequencing technologies deliver millions of sequences per run and make cloning and related generation of plasmid libraries unnecessary.
Common to all second-generation sequencing approaches, however, is the need for construction of DNA libraries through ligation of short adapters, and for these libraries to undergo PCR amplification prior to sequencing (Shendure and Ji 2008). Library building is known to introduce substantial levels of nucleotide misincorporations toward the ends of the reads, most probably as a result of the presence of single-stranded 5′ overhanging ends in DNA templates, which enhances susceptibility to cytosine deamination (Briggs et al. 2007; Brotherton et al. 2007). In addition, primer extension capture of aDNA libraries has shown a significant correlation between read depth and nucleotide composition (GC-rich regions being shorter and over-represented), suggesting that AT-rich sequences might be preferentially lost during library preparation (Briggs et al. 2009). Furthermore, except for keratinous tissues that provide an environment mostly isolated from microbial contamination (Gilbert et al. 2007b, 2008; Miller et al. 2008; Willerslev et al. 2009; Rasmussen et al. 2010) and for some notable exceptions (Poinar et al. 2006; Reich et al. 2010), most aDNA extracts have shown extremely poor endogenous sequence contents (at best 1%–5% of all reads generated), making shotgun sequencing cost ineffective unless DNA capture methods or enzymatic restriction of the microbial fraction are implemented (Briggs et al. 2009; Burbano et al. 2010; Green et al. 2010). Such low ratios of endogenous sequences likely reflect the presence of DNA derived from microbial communities living within the soil, and thus permeating through the fossils; however, as DNA damage and cross-links could hamper adapter ligation and/or library amplification, the low fraction of endogenous DNA may also reflect a bias inflicted by the preferential PCR amplification of undamaged modern contaminant DNA molecules in the steps prior to sequencing.
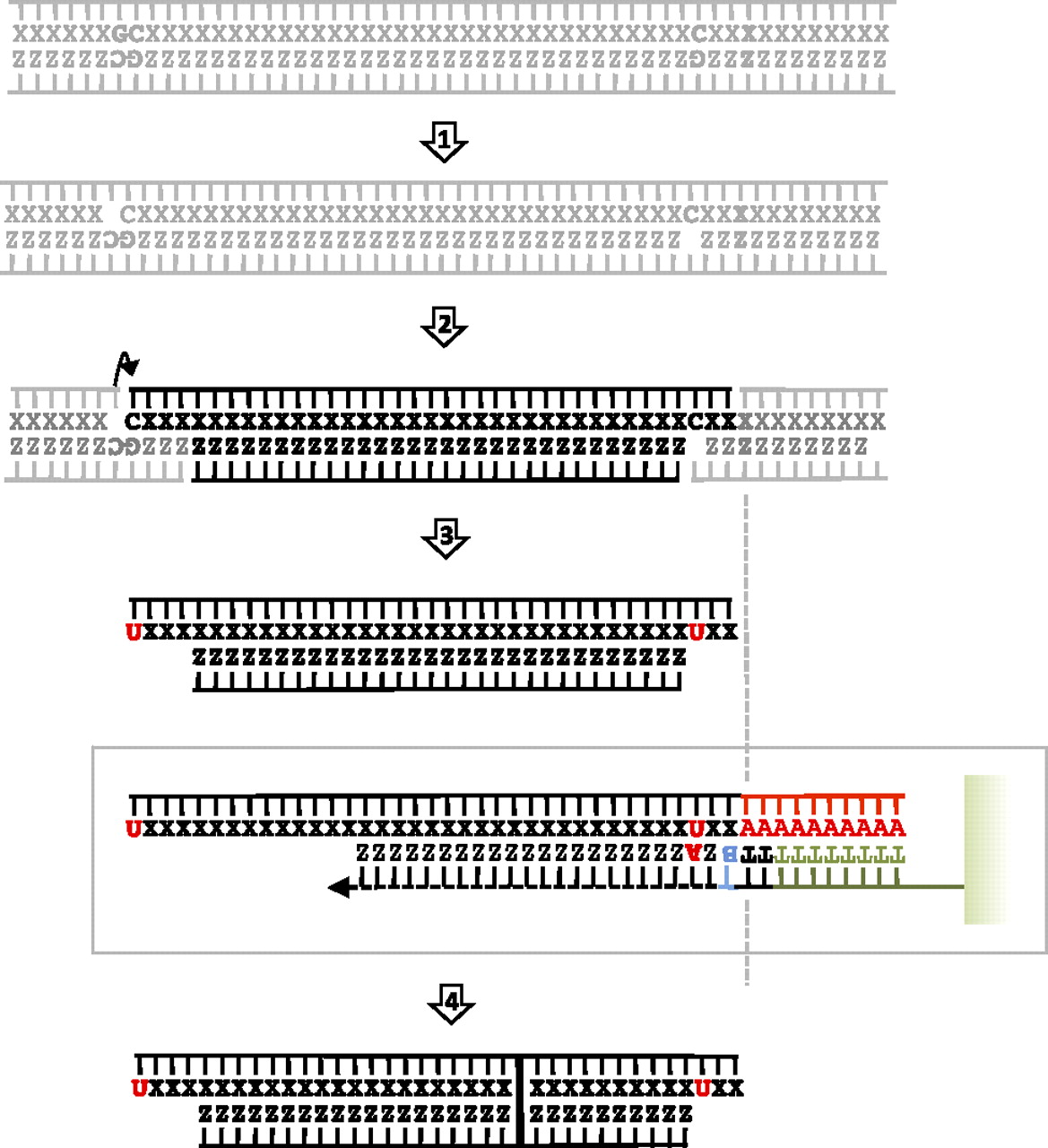
Unlike second-generation sequencing, the so-called “true single-molecule sequencing” techniques (tSMS; alternatively called third-generation sequencing technologies) provide the sequence of single, original template molecules of DNA, avoiding the need for library preparation and amplification (Harris et al. 2008). The HeliScope Sequencer (Helicos BioSciences Corporation) is the first commercially available third-generation platform and currently sequences in a 50-channel format that can deliver up to 30,000,000 reads per channel (Metzker 2010; Thompson and Steinmann 2010). Instead of undergoing the end-repair, ligation, and amplification process, template material is polyadenylated at the 3′ end and captured on a flow cell coated with oligo-dT50. After capture, the template DNA is sequenced by cyclic extension with fluorescently labeled nucleotides (Fig. 1; Thompson and Steinmann 2010). This sequencing technology requires far less material than second-generation technologies and could provide the first massive, direct, and unbiased access to every single molecule preserved in fossils, potentially characterizing the full range of DNA damage through the analysis of nucleotide misincorporation and fragmentation patterns (Stiller et al. 2006; Briggs et al. 2007; Brotherton et al. 2007; Gilbert et al. 2007a; Krause et al. 2010b).
Helicos tSMS: an overview (adapted from Hart et al. 2010 and reprinted with permission from Elsevier Ltd. © 2010). Ancient DNA molecules are denatured into single strands (step 1), tailed with poly(A) (step 2), and captured by oligo-dT-50 oligonucleotide probes covalently linked onto the surface of 25-channels flow-cell (step 3). A fill-in reaction is elicited with dTTP in order to fill any remaining nucleotide complementary to the poly(A) tail (step 4). Nucleic acid templates are then locked in place by the addition of dCTP, dGTP, and dATP virtual terminator (VT, here labeled B) nucleotides that inhibit extension prior to terminator cleavage (step 5). Sequencing-by-synthesis is initiated through the addition of one of the four one-color Cy-5 labeled VT nucleotide (step 6). The incorporation of fluorescence to the elongated DNA strand is measured using laser illumination and a CCD camera after unincorporated nucleotides have been rinsed. The fluorescent label is further cleaved and the incorporation of another labeled VT nucleotide is challenged. Standard sequencing runs complete 120 cycles of nucleotide additions. Ancient DNA, which is extremely fragmented, does not require further shearing before poly(A) tailing.

In this study, we explore the potential of tSMS for sequencing aDNA by contrasting the respective performance of Illumina GAIIx and Helicos HeliScope platforms in terms of sequence yield, relative endogenous sequence content, and DNA damage with the same aDNA extract. This technique enables us to obtain direct access to the 3′-ends of aDNA templates with no need for prior 3′-exonuclease treatment, revealing a new type of structure in ancient molecules, namely, the presence of 3′-overhanging termini.
Results
Overall sequence yields
We generated 330.3 million sequencing reads in this study, using seven GAIIx Illumina lanes (173.0 million reads, 7/8 of a full run) and 12 HeliScope channels (157.3 million reads, ∼1/4 of a run) (Table 1). The majority of the reads did not show any significant sequence similarity to the horse reference genome, with an average of only 1.45%–1.54% of the sequences mapping successfully (Table 1). This is characteristic of large-scale shotgun sequencing of other ancient mammalian bones that have reported ratios ranging from 1% to 5% (Ramirez et al. 2009; Green et al. 2010), with the notable exception for exceptionally well permafrost-preserved mammoth bone (>45%) (Poinar et al. 2006; Miller et al. 2008) and one hominoid phalanx originating from the Denisova cave in the Altai mountains, Southern Siberia (∼70%) (Reich et al. 2010). A significant fraction (17.5%) of the unmapped reads could be identified as environmental bacteria using MegaBlast. Pseudomonadales, a gamma-proteobacteria order including many ubiquitous soil species, was the main bacterial order with 13.9% of the sequences. Other bacterial orders with soil representatives could be identified (Burkholderiales, 0.8%; Actinomycetales, 0.6%), but most of the reads did not show any known close representative and were left unclassified (80.5%). Interestingly, a fraction of sequences showed significant sequence similarity to human sequences, with 0.2% of MegaBlast hits assigned as human and 0.4% of the total number of reads mapping against the genome reference, hg19, suggesting low human contamination levels from excavation to sequencing. Such reads with possible human origin were filtered out from further analyses, even in cases where a higher match against the horse reference genome (eqCab2) was observed.
Illumina versus Helicos tSMS: Overall sequence yields
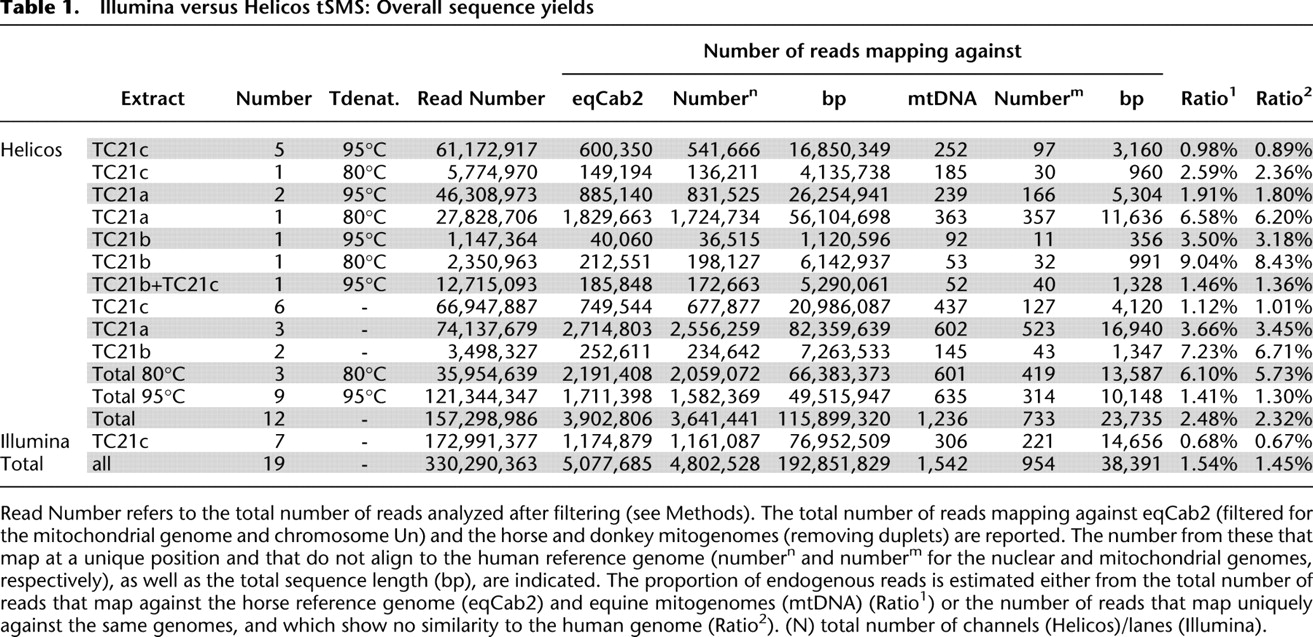
One of the most striking findings was that a higher proportion of the data generated using the Helicos tSMS aligned to the horse genome compared with that generated using the GAIIx, with 1.01%–1.12% and 0.67–0.68% of the total number of sequences, respectively (Table 1). Importantly, these data were generated from the same extract (TC21c; one-way ANOVA with repeated measures, P < 0.0023, excluding data generated at 80°C, which shows even higher endogenous sequence content; see below). However, one lane of Illumina reads covered, on average, 11.0 Mb of the horse genome, whereas only 9.7 Mb of coverage was obtained per Helicos channel (Tables 1, 2). This is due both to the shorter size of Helicos reads (Supplemental Fig. 1) and the relative heterogeneity in the number of reads provided per Helicos channel, but this situation could be improved using a mild denaturation temperature of 80°C in the Helicos template preparation procedure (22.1 Mb of unique horse sequences were recovered per lane at 80°C, in contrast to 5.5 Mb at 95°C; see below).
Contrasting lllumina and Helicos performance and costs for sequencing bone DNA extracts from the Pleistocene
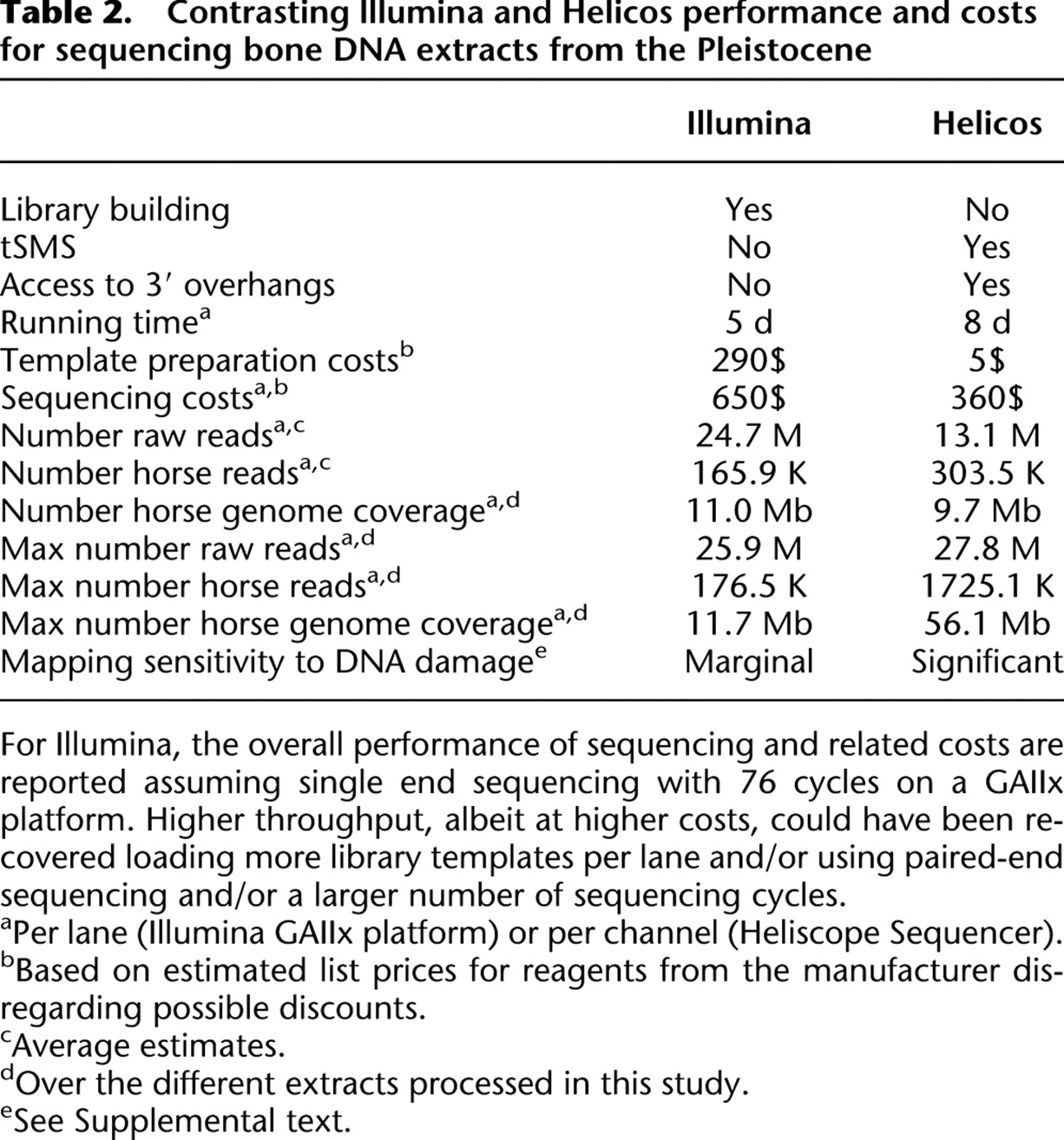
On average, Helicos and Illumina technologies provided similar estimates of the number of mitochondria per cell, with, on average, one mitochondrial read being observed every 4968 and 5254 nuclear reads, respectively, which is in the range of what has been reported for permafrost-preserved mammoth bones based on shotgun sequencing (658 in Poinar et al. 2006) or real-time PCR measurements (245–17,480) (Schwarz et al. 2009). Given the respective sizes of the horse mitochondrial and nuclear genomes (16,660 bp and 2.37 Gb) and assuming similar size distribution for nuclear and mitochondrial reads, this indicates that ∼54–58 mitochondria per cell (nucleus) are preserved in the bone material analyzed. The similarity between sequencing approaches suggests that no bias toward any particular genome type was introduced during library preparation and amplification required by Illumina sequencing. This is further supported by the balanced distribution of reads over the different nuclear chromosomes, with significant correlation between the number of mapped reads and chromosome size (Pearson correlation coefficient >0.935, P < 5.1 × 10−15; Supplemental Fig. 2).
The higher fraction of endogenous sequences present in Helicos data indicates that the Illumina sequencing recovered proportionally more environmental DNA sequences. This is further reflected by MegaBlast results on Helicos reads that show a 2.7-fold decrease in bacteria hits compared with Illumina reads (10.8% vs. 28.9%), while the fraction of unassigned hits only increases 1.3-fold (87.3% vs. 69.0%). DNA damage present in aDNA molecules may interfere with end-repair reactions, ligation, and amplification, resulting in relatively lower endogenous sequence yields when these steps are required. Whether this bias is introduced during library preparation, library amplification, or a combination, remains to be determined. Of note, the Helicos and Illumina sequence reads show similar base compositions, with average GC content (44.4% and 44.9%) equally distant to the expected value observed for randomly sampled genomic fragments of similar size (41.4%) (Fig. 2). Overall, this suggests that tSMS approaches are able to characterize a larger fraction of aDNA extracts by accessing more endogenous molecules; however, in contrast to previous reports that have shown under-representation of AT-rich regions (Hillier et al. 2008; Quail et al. 2008), the procedure followed here for Illumina sequencing has not introduced substantial bias in read base composition.
GC composition of Illumina and Helicos horse reads. For comparison, we considered only the reads generated from the same extract (TC21c) and denaturation temperatures of 80°C and 95°C. Similar distributions were recovered when considering the total number of Helicos reads generated for other extracts. (Left) Helicos; (right) Illumina. Full lines refer to the observed average read GC content. The expected average GC content of genomic fragments of 31 bp (Helicos read median) is estimated using 361,379 randomly sampled fragments of the horse reference genome (see Supplemental text) and is reported in dashed lines (41.41%). A similar estimate (41.38%) is provided for Illumina sequencing reads using 299,256 randomly sampled fragments of 67 bp, in agreement with the median of Illumina sequences.

Ancient DNA content of different bone fractions
Three independent DNA extracts from the horse bone were sequenced on the HeliScope Sequencer. One, TC21c, was generated from fresh bone powder following complete digestion in an EDTA-rich decalcifying buffer (see Methods). The other two extracts, TC21a and TC21b, consisted of re-extraction of undigested pellets from a previous extraction of some bone powder originating from the same bone specimen. The latter two are therefore more representative of DNA molecules preserved in demineralized and undigested bone particles, while the former includes contributions of the mineralized and collagen-rich fractions.
The three types of extracts delivered substantial amounts of horse sequence data, confirming that some, but not all aDNA molecules were released in the first extraction round, confirming previous reports (e.g., see Schwarz et al. 2009). Of note, extracts TC21a and TC21b also show relatively longer read length with median sizes superior to TC21c regardless of the template preparation protocol used for Helicos sequencing (denaturation at 80°C or 95°C) (Fig. 3, left). The size distribution of Helicos reads does not correspond to the size distribution of aDNA templates as most sequencing-by-synthesis reactions do not reach the end of the molecules. However, it is likely that many of the sequence reads are full length, as standard read length observed on fresh DNA is longer than observed with aDNA. This observation is compatible with the presence of longer molecules in extracts coming from undigested bone particles (TC21a and TC21b). This is further confirmed by the purine content of Helicos reads (e.g., the class of reads showing a %GA > 60.0%), which decreases as a function of sequence length. This suggests that for a fraction of the reads (Supplemental Fig. 3), the sequencing-by-synthesis reaction stops at depurinated sites, in agreement with models of DNA fragmentation through depurination (Briggs et al. 2007). Longer DNA templates appear to have been conserved in the demineralized and undigested pellets after the first round of extraction, confirming previous hypotheses that the enrichment in short fragments resulting from bone demineralization could be due to size filtration of DNA templates through the collagen matrix, releasing preferentially short molecules in decalcifying buffers (Schwarz et al. 2009).
The distribution of Helicos reads is dependent on the initial denaturation temperature. Three different extracts (top: TC21c; middle: TC21b; bottom: TC21a) have been sequenced on the same Helicos run (six channels) following identical procedures, except that either mild (80°C, black) or high (95°C, gray) temperatures were used for denaturation. (Left) Read length distribution. For extracts TC21c, TC21b, and TC21a, the median read size was 29, 30, and 32 bp when DNA denaturation was performed at 80°C (black dashed lines) in contrast to 27, 29, and 29 bp at 95°C (gray dashed lines). (Middle) Read GC contents. White full lines refer to average read GC contents; the expected genomic GC content (41.4%) is reported with dashed lines. (Right) Cumulative guanine to adenine misincorporation rates as a function of the distance from sequencing start.

Importantly, the three types of extracts differed in endogenous sequence contents. Compared with TC21c, the re-extracts, TC21a and TC21b, were enriched in horse sequences relative to the overall number of reads (3.3-fold to 6.6-fold) (Table 1). Whether this observation is specific to the specimen analyzed or characteristic of ancient mineralized tissues in general needs further investigation. However, this effect was replicated in an additional extraction and re-extraction experiment performed on different pieces of the same horse bone as well as on another permafrost-preserved horse fossil bone (data not shown). These results suggest that the first extraction round may preferentially wash out exogenous environmental DNA, while leaving substantial amounts of endogenous DNA molecules entrapped in the undigested pellets. These pellets will therefore represent relatively contamination-free niches for DNA preservation, similar to the crystal aggregate hypothesis of Salamon et al. (2005).
Improving endogenous sequence yields
Post-mortem chemical alterations result in extensive fragmentation and modification of DNA molecules (Paabo 1989). The standard Helicos sequencing protocol is initiated with a DNA denaturation step at 95°C in order to generate single-stranded DNA for terminal transferase tailing. As ancient DNA fragments are short, damaged, and exhibit substantial levels of overhanging ends (Briggs et al. 2007), they may be more prone to denaturation at mild temperatures (80°C) than modern contaminant DNA templates. Hence, mild denaturation temperatures could improve endogenous sequence yields. In addition, high denaturation temperatures might further increase fragmentation and/or deamination in aDNA templates, leading to shorter reads and/or higher levels of nucleotide misincorporations. To investigate this, we compared the results from the same sequencing run on the HeliScope Sequencer of two preparations of each the three extracts (TC21a, TC21b, and TC21c), in which similar volumes of each extract were denatured at different temperatures (80°C and 95°C).
Strikingly, for all pairs, the fraction of endogenous horse sequences was higher (2.6-fold to 3.4-fold) after initial denaturation at 80°C than at 95°C, with no apparent reduction in the total number of sequences recovered per channel (Table 1). Overall, 49.5 Mb of horse sequences were identified using nine channels and 95°C as a denaturation temperature, while 66.4 Mb were generated out of only three channels when denaturation was performed at 80°C (Table 1). In addition, read size distributions were shifted upward at the lower denaturation temperature (Fig. 3, left), suggesting that higher denaturation temperatures, even for short incubation steps, may enhance DNA fragmentation through the formation of single-strand breaks.
For all extracts, horse reads recovered from 80°C denaturation treatments exhibited lower GC contents than reads recovered from the 95°C treatment (Fig. 3, middle). In addition, higher guanine to adenine misincorporation rates were observed within the double-stranded part of aDNA molecules (see below), with cumulative rates over nucleotide positions 4–25 ranging from 39.3% to 41.0% at 80°C vs. 33.6% to 38.8% at 95°C (Fig. 3, right). As the deamination of cytosine to uracil results in the loss of one hydrogen bond in every deaminated GC pair, we believe that mild temperatures slightly favored the denaturation of ancient deaminated templates, both reducing read GC contents (uracils are analogs of thymines) and increasing the fraction of endogenous (damaged) sequences recovered (hence, the rate of guanine to adenine misincorporation).
Interestingly, in TC21a and TC21b extracts, the level of nucleotide misincorporation observed at the 3′-ends of aDNA templates was found to increase in reads generated from the 80°C denaturation procedure, with cumulative G-to-A substitution rates of 29.3% and 25.6% along the first three nucleotides sequenced (compared with 23.1% and 21.4% at 95°C, respectively). The reverse was found for extract TC21c with G-to-A substitution rates of 25.5% and 32.9%, respectively (Fig. 3, right). This suggests that in addition to mild denaturation temperatures that deliver higher proportions of endogenous sequences, complete bone demineralization and digestion provide access to a fraction of aDNA templates with relatively lower deamination at 3′-ends. In such extracts, aDNA templates with shorter overhanging ends and higher cytosine deamination in double-stranded regions were made preferentially available for tSMS by denaturing at 80°C; reversely, denaturation at 95°C provided preferential access to templates with longer 3′-overhang termini (Fig. 3, right), but lower cytosine deamination in double-stranded regions, as shown by higher GC content (Fig. 3, middle). In contrast, undigested bone pellets represent a relatively contamination-free niche of relatively longer DNA molecules; for such molecules, the energy provided by a mild temperature is compatible with preferential denaturation of templates with longer single-stranded overhangs and higher cytosine deamination levels; high denaturation temperatures (here, 95°C) provide access to most of aDNA templates, including those with shorter overhangs and lower deamination levels (Fig. 3, right).
The observation that endogenous sequence yields can be increased using mild denaturation temperatures has important consequences for genome-wide surveys of ancient organisms, as it makes it feasible to recover more ancient sequence reads both from fewer sequencing runs and less DNA extract. As most extraction procedures are destructive, the latter could be critical when material sources are scarce. tSMS of ssDNA templates denatured at mild temperatures therefore provides an alternative to library-enrichment procedures such as in-solution DNA capture (Briggs et al. 2009; Maricic et al. 2010), micro-array capture (Burbano et al. 2010), and enzymatic restriction of bacterial DNA (Green et al. 2010).
DNA damage
Illumina sequencing
Ancient DNA sequence reads typically show increased levels of nucleotide misincorporation at both ends (Briggs et al. 2007). In particular, cytosine-to-thymine and guanine-to-adenine nucleotide misincorporations appear preferentially at the 5′- and 3′-termini of sequences, respectively (Briggs et al. 2007; Brotherton et al. 2007; Krause et al. 2010b). This is most likely due to the presence of 5′-overhanging ends, as single-stranded DNA exhibits faster rates of cytosine deamination than does double-stranded DNA (Lindahl 1993). Furthermore, the base composition of the nucleotide preceding the first nucleotide sequenced at the 5′-end of the aDNA reads shows elevated levels of purines; a symmetric excess in pyrimidines has been found at 3′-ends (Briggs et al. 2007), suggesting that depurination is a key component of post-mortem fragmentation of aDNA molecules.
The Illumina reads identified as endogenous exhibit both DNA degradation features; of note, modern human reads showed neither the nucleotide misincorporation nor the DNA fragmentation patterns observed in the horse reads (Supplemental Fig. 4), confirming that overall patterns of DNA degradation can be used to distinguish genuine endogenous DNA sequences from modern contaminants (Krause et al. 2010ab). Cytosine to thymine misincorporation rates are highest (∼30.7%) at the first position of the sequences and decrease by approximately twofold per position as the read progresses (Fig. 4, bottom). This rate was reduced to 3.2% at the fifth nucleotide. A symmetric situation was observed at the 3′-end, except that guanine-to-adenine transitions, instead of cytosine-to-thymine, are detected. In addition, this misincorporation occurred at lower rates (∼25.1% for the last nucleotide position in sequence reads), suggesting that a substantial fraction of the sequences did not reach the end of the aDNA template, and that further sequence information could have been gained by extending the number of sequencing cycles from 76 to 100 or by performing paired-end sequencing. We note that ∼4000-yr-old human hairs preserved in the permafrost exhibited a 9.2-fold decrease in cytosine-to-thymine misincorporation rate at the first position of Illumina reads (∼3.3%) (Ginolhac et al. 2011). With deamination levels superior to the one observed from ∼40-KY-old neandertal bone specimens excavated from a temperate cave (∼22% at the first position of sequencing reads) (Briggs et al. 2007), the permafrost-preserved horse specimen analyzed here could be much older than 40 KY, in agreement with its infinite radiocarbon age.
Illumina sequencing: DNA fragmentation and nucleotide misincorporation patterns on ancient horse reads. (Top, middle) The base composition of the reads is reported for the first 10 nucleotides sequenced (left: 1–10) as well as for the five nucleotides located upstream of the genomic region aligned to the reads (left: −5 to −1). In addition, the base composition of the last 10 nucleotides sequenced (right: −10 to −1) and of the five nucleotides located downstream from the reads (right: 1–5) in the genome equCab2 is provided. Nucleotide positions located within reads are reported with a gray frame. Each dot reports the average base composition per position as estimated from reads mapping against chromosomes 1–31 and X. The range of the base composition per individual chromosome is also reported. (Bottom) The frequencies of all possible mismatches and indels observed between the horse genome and the reads are reported in gray as a function of distance for 5′- to 3′-ends (first 25 nucleotides sequenced) and 3′- to 5′- (last 25 nucleotides), except for C→T and G→A, which are reported in red and blue, respectively. The latter variations range from 0.6% to 30.7% per site (5′- to 3′- end) or 0.7%–25.1% per site (3′- to 5′- end) and exceed the variations observed for other misincorporation types that are consequently mostly hidden in the figures (<0.1%–0.9% per site). The misincorporation frequencies are calculated by dividing the total number of occurrences of the modified base at a given position in a read by the total number of the unmodified base at the same position in the horse genome.
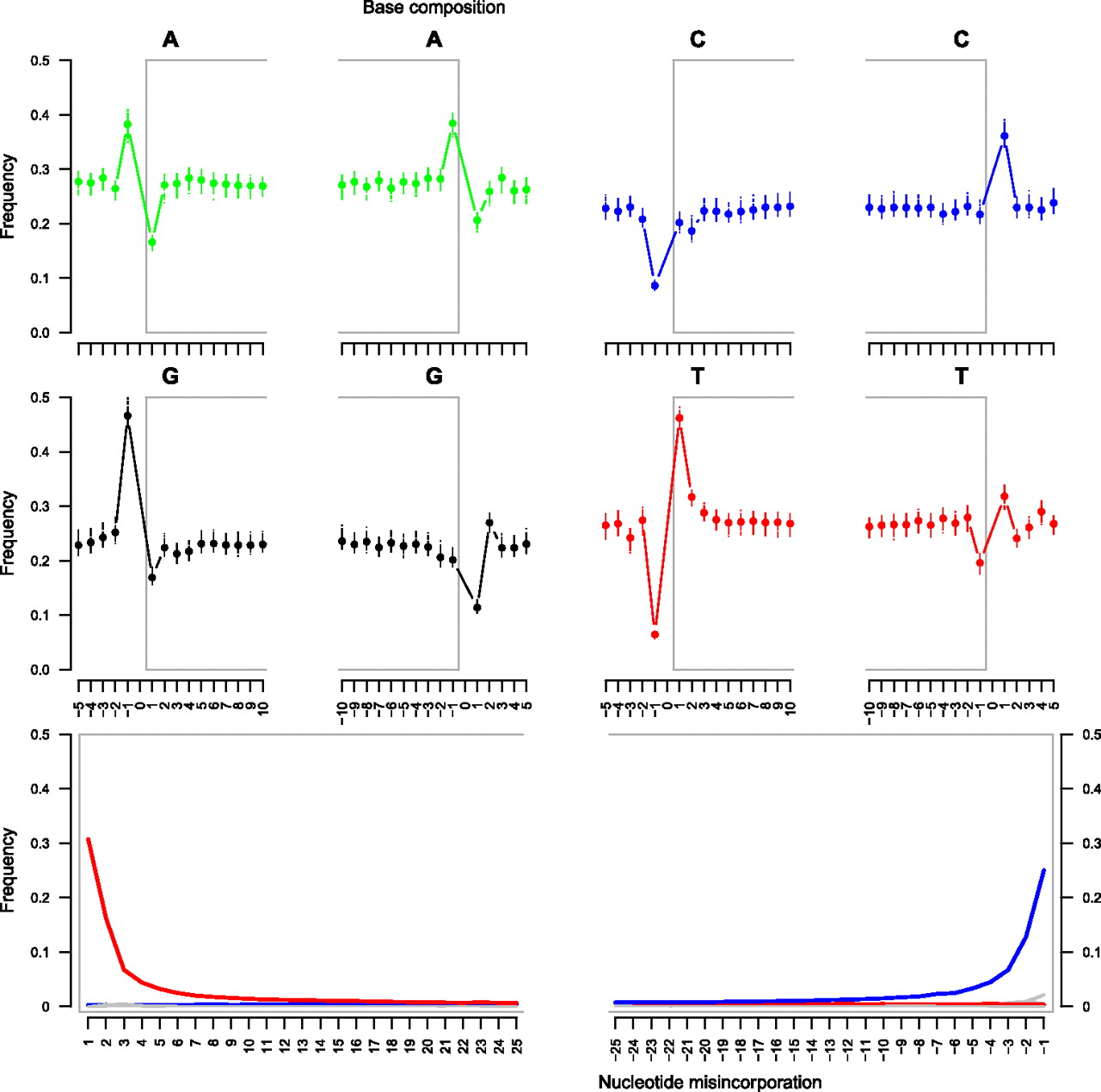
Excessive proportions of purines (or pyrimidines, respectively) were detected in the genomic region located 5′ (or 3′) of sequence reads, but these were limited to the nucleotide position preceding (following) sequencing starts (ends), confirming the model of DNA fragmentation through depurination (Fig. 4, top and middle). Interestingly, between purines, guanines were the most affected, suggesting that abasic sites appeared at higher rates post-mortem at guanine relative to adenine sites. The excess in pyrimidines observed at the 3′-end of the sequences (from 21.7% to 36.1% and from 19.6% to 31.8% for cytosine and thymine residues at the last position sequenced and the following nucleotide position in the reference genome) (Fig. 4, top and middle) is not equal to the excess in purines detected at the 5′-end (from 16.6% to 38.2% and from 16.9% to 46.6% for adenine and guanine residues, respectively) (Fig. 4, top and middle). This is reminiscent of the nucleotide misincorporation pattern and again indicates that a substantial fraction of the sequences did not reach the end of the aDNA template.
Helicos sequencing
During Helicos sequencing template preparation, DNA molecules undergo denaturation, poly(A) tailing, blocking, and oligo-dT capture (Fig. 1). These steps could introduce bias in the population of molecules sequenced and may affect both patterns of nucleotide misincorporation and DNA fragmentation. We therefore characterized these possible sources of bias using available Helicos sequence data generated from modern human genomic sequences (Pushkarev et al. 2009). We identified three typical features in the nucleotide composition of the genomic regions sequenced (see Supplemental text). First, post-sequencing trimming of the reads when starting with thymine residues resulted in the absence of thymine in the first position of sequence reads (Supplemental Fig. 5). Second, dGTP virtual terminator residues are preferentially incorporated during the locking reaction (Fig. 1), resulting in guanine enrichment in the genomic coordinate located just before the sequencing start (Supplemental Fig. 5). One consequence of post-sequencing read trimming and preferential locking efficiency with dGTP virtual terminators is that the first nucleotide sequenced is not necessarily the next to last nucleotide of the aDNA strand (the last corresponding to the site locked) (Supplemental Fig. 6), but might be located a few nucleotides further into the preserved molecule. The third deviation to the average nucleotide composition consisted of a progressive excess in thymine residues in the genomic region preceding the blocking site (Supplemental Fig. 5), suggesting that adenine-rich templates outcompete other genomic regions during the oligo-dT capture step (Fig. 1).
Interestingly, all three biases, except the excess in guanine residues at the locking sites, are observed on the human reads generated from the ancient horse extracts (Supplemental Fig. 7). Of note, the first nucleotide sequenced showed extremely low, but not null frequencies in thymine residues, as sequences starting with a minimum of two (and not one) thymine residues were trimmed post-sequencing (Supplemental Fig. 6).
The deficit in thymine residues detected at the first position within sequence reads also affected the frequency of nucleotide misincorporation (Supplemental Fig. 8). At that position, thymine residues are trimmed unless a misincorporation is observed, resulting in rate estimates ranging from 26.1% to 45.5% per base for thymine-to-guanine and adenine misincorporations, respectively (Supplemental Fig. 8). This pattern was found to be less pronounced on the human reads generated from the ancient horse extracts (Supplemental Fig. 9) due to the less strict rules used for trimming. Except deletions that dominate error types with average rates of 2.1% per base (range: 0%–3.7%), all other misincorporation types were observed at much lower rates all along the sequence position both in the published human reads (Supplemental Fig. 8) and the human reads generated from the ancient horse extracts (Supplemental Fig. 9). Such high occurrence of deletions most likely reflect nucleotide incorporation without detection in the course of Helicos sequencing, which is characteristic to tSMS (Bowers et al. 2009; Thomson and Steinmann 2010).
We next investigated the DNA fragmentation and nucleotide misincorporation patterns for aDNA using the ancient horse reads (Fig. 5, top and middle). As Helicos sequencing starts from the 3′-end of the aDNA strands, this gave us a unique opportunity to document the chemical nature of the 3′-termini in aDNA molecules. As expected from the post-sequencing trimming procedure, we observed a deficit in thymine residues and parallel excess in adenine and cytosine residues at the first position of sequence reads; no excess in guanine residues was observed due to the high rates of guanine-to-adenine misincorporation at the first nucleotide position of Helicos reads (see below). Additionally, the blocking site showed preference for guanine residues, as observed in modern human sequence reads. Furthermore, we found the expected progressive increase in thymine residues in the genomic region preceding the blocking site. On fresh DNA templates, however, this was paralleled by a progressive decline in adenine as well as cytosine and guanine residues (Supplemental Fig. 5). While we found evidence of a similar decline in adenine residues, cytosine and guanine composition showed subtle differences, with a slight enrichment in cytosine residues, which slightly enhances the expected deficit in guanine residues (Fig. 5, top and middle; Supplemental Fig. 5).
Helicos sequencing: DNA fragmentation and nucleotide misincorporation patterns on ancient horse reads. (Top, middle) The base composition of the reads is reported for the first 10 nucleotides sequenced (left: 1–10) as well as for the five nucleotides located upstream of the genomic region aligned to the reads (left: −5 to −1). In addition, the base composition of the last 10 nucleotides sequenced (right: −10 to −1) and of the five nucleotides located downstream from the reads (right: 1–5) in the genome equCab2 is provided. Nucleotide positions located within reads are reported with a gray frame. Each dot reports the average base composition per position as estimated from reads mapping against chromosomes 1–31 and X. The range of the base composition per individual chromosome is also reported. (Bottom) The frequencies of all possible mismatches and indels observed between the horse genome and the reads are reported in gray as a function of distance for 5′- to 3′-ends (first 25 nucleotides sequenced) and 3′- to 5′- (last 25 nucleotides), except for C→T, G→A, deletions, and insertions that are reported in red, blue, green, and pink, respectively. These frequencies are calculated by dividing the total number of occurrences of the modified base at a given position in a read by the total number of the unmodified base at the same position in the horse genome. For indels, the latter corresponds to the total number of bases observed at the considered position.

Although high occurrences of deletions were observed as expected for tSMS (on average 3.2% per base; range: 0%–5.4% per base), the nucleotide misincorporation pattern observed in the ancient horse reads was strikingly different from that observed with modern DNA (Fig. 5). In particular, the most frequent substitution was from guanine to adenine residues, confirming that cytosine deamination to uracil is the most important driver for nucleotide misincorporation (Paabo 1989; Hansen et al. 2001; Hofreiter et al. 2001; Gilbert et al. 2003, 2007a; Stiller et al. 2006; Briggs et al. 2007; Brotherton et al. 2007). In the sequencing-by-synthesis reaction, the uracil in the aDNA strand is recognized as a thymine residue, giving rise to the incorporation of an adenine instead of the guanine expected if the cytosine was not deaminated (Supplemental Fig. 10). More importantly, while it was more common than all other substitution types at all nucleotide positions within the read, the rate of guanine-to-cytosine deamination showed a marked increase in the first three nucleotides sequenced, with a nearly twofold increase (1.8×) from the third position to the second (from 4.5% to 8.3% per base), and 1.7× from the second to the first (from 8.3% to 14.0% per base). Identical patterns are found even if indels are allowed in the first five positions of mapped reads (Supplemental Fig. 11A) or disallowed in the first 10 positions (Supplemental Fig. 11B), suggesting that the detected pattern is not a misalignment by-product. Reminiscent of what has been observed at the 5′-end of Illumina reads, the pattern of guanine-to-adenine misincorporations is most probably due to the presence of 3′-overhanging ends in aDNA templates, as single-stranded DNA exhibits faster rates of cytosine deamination than double-stranded DNA. Bearing in mind that the first nucleotide sequenced in Helicos reads is not the last, but at best the penultimate nucleotide of the aDNA strand (Supplemental Figs. 6, 10), Illumina and Helicos reads provided rather similar cytosine deamination rates in 5′- and 3′-overhanging ends, as expected for single-stranded regions (at the second position of Illumina reads cytosine to thymine misincorporation rates of 16.2% per base were observed, which is similar to the 14.0% observed at the first position of Helicos reads for complementary guanine-to-adenine misincorporations).
Interestingly, we found similar cytosine deamination rates in the mitochondrial and nuclear genomes, suggesting that both genomes exhibit similar deamination trajectories post-mortem (χ2 test, P = 0.9105 and 0.0659 considering either the first position of sequencing reads or all positions).
Discussion
Post-mortem DNA damage
The occurrence of post-mortem damage in DNA extracted from fossil remains has been recognized as a longstanding problem for analyzing aDNA molecules (Paabo 1989). Oxidative derivatives of pyrimidines, abasic sites, and intermolecular cross-links have been shown to preclude molecular cloning, restricting its efficiency to exceptional cases only (Paabo et al. 1989; Lindahl 1993), unless end-repair reactions with DNA polymerase and PNK activities were performed (Noonan et al. 2005, 2006). While still sensitive to polymerase blocking lesions, such as abasic sites, single-strand breaks and intermolecular cross-links, PCR has opened access to a wide variety of aDNA templates coming from different preservation environments (e.g., Antarctic coast [de Bruyn et al. 2009] or Balearic islands [Ramirez et al. 2009]), source material (e.g., bones [Green et al. 2010] or egg shells [Oskam et al. 2010]) and time periods (10,000–500,000 yr ago) (Willerslev et al. 2004). However, another type of damage, namely miscoding lesions, promotes nucleotide misincorporation during PCR amplification of DNA fragments extracted from ancient organisms (Paabo et al. 1989), the most important consisting of the deamination of cytosine into uracil, which leads to G/C to A/T misincorporation (Hansen et al. 2001). Amplicons showing substantial levels of misincorporation have even been shown to represent a significant proportion of PCR products when only a few templates are available for amplification, generating false sequence information, and, hence, requiring independent sequence validation from multiple PCR products and clones (Hofreiter et al. 2001).
Massively parallel sequencing approaches have prompted a revolution in the characterization of aDNA by providing access to the nuclear genome (Poinar et al. 2006; Miller et al. 2008; Green et al. 2010; Rasmussen et al. 2010; Reich et al. 2010). In addition, these second-generation technologies have lead to a better understanding of the process of post-mortem DNA damage by delivering hundreds of millions of sequences (Stiller et al. 2006; Gilbert et al. 2007a). This revealed that depurination was a driving force of DNA fragmentation post-mortem, as an excess of purines has been detected in the genomic position preceding the starts of reads generated using 454 sequencing (Briggs et al. 2007). Furthermore, the 5′-termini of reads are found to be enriched in cytosine-to-thymine misincorporations. This suggested the existence of 5′-overhanging ends in a substantial fraction of aDNA molecules, as the rate of cytosine deamination into uracil is much faster in single-strand DNA than double-strand DNA (Lindahl 1993). At 3′-ends, complementary misincorporations (guanine-to-adenine) are found as a result of the fill-in reaction during library construction, with the incorporation of adenine, and not guanine residues at sites showing cytosine deamination (Briggs et al. 2007). These patterns have been later confirmed using larger Illumina sequence data sets and ancient human extracts from the late Pleistocene (Briggs et al. 2009; Krause et al. 2010a,b). The permafrost-preserved horse specimen analyzed here shows similar nucleotide misincorporation and DNA fragmentation patterns. The fact that none of these patterns could be found in contaminating human (Supplemental Fig. 4) and Pseudomonales-related sequence reads (data not shown) confirms that DNA damage could be used as a proxy for aDNA authenticity, especially in cases when results are at a particular risk from contamination (e.g., ancient human extracts) (Wall and Kim 2007; Krause et al. 2010a,b).
The Helicos tSMS reads generated in this study, which do not require DNA copying prior to sequencing, have confirmed that adenine residues are most often misincorporated instead of guanine residues when sequencing aDNA templates, most probably as a result of cytosine deamination in the original aDNA strand. Furthermore, as the poly(A) tailing reaction starts from the 3′-termini of the ancient template, Helicos tSMS provides a unique opportunity to characterize the 3′-end of aDNA templates, with no need for end-repair beforehand (Fig. 1). With library-dependent sequencing approaches, including Illumina, any 3′-overhang possibly present in aDNA templates is processed by the 3′ to 5′ exonuclease activity of the T4 DNA polymerase, and therefore remains inaccessible to sequencing. Yet, in Helicos reads, guanine-to-adenine misincorporations decrease progressively as the distance from the 3′-end increases (Fig. 5). This is reminiscent of the cytosine-to-thymine misincorporation pattern observed in Illumina reads at the 5′-ends and suggests the existence of 3′-overhanging ends in aDNA templates. Although terminal transferase has shown reduced affinity for adding tails to modified bases (data not shown), the high cytosine-to-thymine misincoporation rates observed here suggest that terminal transferase activity is not particularly sensitive to deaminated nucleotidic bases found at the 3′-ends of aDNA templates, and that the tailing does not bias the available pool of molecules available for sequencing through preferential tailing of unmodified templates.
Furthermore, comparison of Helicos reads generated from modern human DNA, with the results from the horse aDNA, shows no major differences in the base composition of the genomic region preceding sequencing reads, suggesting that 3′-ends in aDNA templates are mainly generated through single-strand breaks occurring at all possible bases (Fig. 6). However, the composition of guanine residues was slightly decreased, suggesting that a minority of the 3′-termini consists of abasic sites formed through loss-of-guanines. At sites of base loss, the DNA chain is weakened and undergoes further cleavage through a β-elimination reaction, leaving DNA fragments with phosphorylated 5′-ends and modified 3′-ends (Lindahl 1993; Mitchell et al. 2005). Helicos tSMS is dependent on efficient poly(A) tailing of free 3′-hydroxyl termini through terminal transferase. Because we see high depurination rates at 5′-ends, we believe that the apparent poor depurination levels observed at 3′-ends of aDNA templates is a by-product of terminal transferase activity, which is unable to tail modified (e.g., phosphorylated) 3′ ends. We believe that both single-strand breaks and depurination at guanine residues appear as the main drivers of DNA fragmentation post-mortem, even though the latter are less compatible with tailing as used in standard Helicos tSMS. This issue could be overcome by further enzymatic treatments (e.g., phosphatase), but at the price of further loss of some of the DNA substrate, a significant problem with trace samples such as these.
Ancient DNA damage: a profile. After depurination (step 1), internal AP-sites are subject to β-elimination (arrow, step 2), which opens the phosphodiester bond mainly for 3′ of AP-sites. In addition, DNA strands are subject to single-strand breaks. As a result of terminal transferase preference for 3′-hydroxy ends, most abasic sites located 3′ of the aDNA fragment will not be poly(A) tailed, unless the nucleotidic sugar is further degraded. Such termini are not represented, albeit they are likely to represent a significant fraction of aDNA templates. Cytosine deamination in uracils occurs much faster on single-stranded parts of DNA (step 3) and results in increased G→A misincorporation rates at the beginning of Helicos sequence reads. Other types of damages, such as interstrand cross-links, which affect aDNA molecules (and hamper further sequence characterization), are not reported.
Sequencing ancient genomes
In this study, we used seven Illumina GAIIx lanes to generate 76.9 Mb of sequence information that could be mapped to a unique region in the horse reference genome. With 11.0 Mb per lane (Table 2), a minimum number of 215 GAIIx lanes (∼27 runs) would be required for covering the 2.37 Gb horse reference genome at 1×. In contrast, 12 Helicos channels provided 115.9 Mb (Table 1), which represents an average performance of 9.7 Mb of horse sequence per channel. However, we demonstrated that at mild denaturation temperatures (here, 80°C), higher endogenous sequence yields could be recovered, yielding up to 56.1 Mb of horse sequence information per single Helicos channel (Tables 1, 2). In this optimal situation, no more than 42 Helicos channels, i.e., less than a full sequencing run, would be needed to generate a draft of the ancient horse genome at 1× coverage.
Moderate denaturation temperatures (here, 80°C) most likely improve the ratio of endogenous to contaminating sequences generated in Helicos sequencing, as aDNA molecules are more prone to denaturation than fresh environmental DNA that derives from microbial communities living in the soil and fossil. Furthermore, moderate denaturation temperatures could limit further DNA degradation as read length slightly decreased after denaturation at 95°C (Fig. 3, left). Since the class of GA-rich sequences was under-represented for longer reads compared with shorter reads, we suggest that the further fragmentation at 95°C could result from depurination (Supplemental Fig. 3), in agreement with rapid DNA depurination rates measured in vitro (Lindahl 1993). This suggests that in addition to the storage conditions of fossil specimens after excavation (Pruvost et al. 2007), experimental procedures associated with DNA sequencing and manipulation could further increase the levels of damage present in aDNA molecules.
Importantly, the extracts analyzed in this study show high levels of depurination and deamination, potentiating even further the damage that could result from high denaturation temperature treatments. Likewise, we could expect the recovery of endogenous sequences to be significantly improved for aDNA extracts showing a similar extent of DNA damage, e.g., material beyond the limits of radiocarbon dating and/or coming from poor preservation environments. However, it is unlikely that DNA denaturation at mild temperatures would improve the ratio of endogenous to exogenous sequences recovered in cases where only moderate DNA damage is present. In addition, we note that the main environmental microbe identified in the extract is closely related to Pseudomomas, a bacteria whose genome exhibits high GC content (e.g., 60.5%–63.4% for P. fluorescens Pfo-1 and Pf-5) (Kimbrel et al. 2010). Mild temperatures would favor the denaturation of horse fragments over Pseudomonas, as the former exhibit much lower GC content (Fig. 2). Again, we could anticipate that DNA denaturation at mild temperatures would not provide higher endogenous sequence yields in cases where the environmental microbial contaminant fraction consists of AT-rich metagenomes and/or the ancient genome under study was GC-rich.
Our finding that endogenous sequence yields can be increased using mild denaturation temperatures might have important consequences for the most widely used method in aDNA analysis, namely the PCR, as repeated denaturation cycles at high temperature could increase DNA fragmentation and reduce the number of templates available in the initial stages of amplification. The probability of jumping-PCR could be expected to increase at the same time as the increasing number of fragments could prime the amplification reaction and generate spurious amplification products (Paabo 1989). For aDNA fragments showing extensive levels of DNA damage, optimal PCR efficiency should therefore be achieved by lowering denaturation temperatures; this would preclude using hot-start DNA polymerases but should not affect PCR specificity unless the reactions are performed under nonstringent conditions.
One aDNA extract, TC21c, was used to generate shotgun sequences on both Illumina GAIIx and HeliScope Sequencer. Interestingly, different ratios of endogenous sequences were recovered, with higher performance observed for tSMS (Table 1). This demonstrates that the overall molecular complexity present in second-generation DNA libraries does not provide an unbiased representation of the sequence complexity originally present in ancient extracts. Whether this difference is introduced during library construction and/or amplification still needs further investigation. However, clonal expansion affected here 10.6% of horse reads, suggesting that a significant bias is introduced during Illumina library amplification. Additional sources of bias have been reported, e.g., due to the size selection step after library amplification as melting gel slice by heating in chaotropic buffer tends to enrich for GC rich sequences (Quail et al. 2008). In the case of the horse genome that shows overall 41.4% of GC content (Fig. 2), this could have resulted in under-representation of endogenous fragments; however, the overall GC content of Helicos and Illumina reads are virtually identical, suggesting that no significant bias in base composition has been introduced during gel purification of the amplified Illumina library, most likely because gel slices have been melted at a moderate temperature (37°C).
Our data suggest that under optimal conditions, on this sample, no more than two Helicos channels (out of 50 per run) would be needed to generate the same amount of horse sequence information as a complete Illumina GAIIx run. We note, however, that a large variation was observed among the different DNA extracts analyzed, both in the overall number of sequences generated per Helicos channel, and in the ratio of endogenous horse sequences (Table 1). This first confirms that the process of DNA preservation is dependent on micro-environmental conditions within fossilized bones, but additionally that large differences in sequence outcomes can occur with tSMS, making global predictions of the amount of material needed difficult. Additional experimental procedures such as extract concentration and oligonucleotide spiking of DNA extracts have been shown to normalize and improve Helicos sequence yields. In fact, on the Helicos Genetic Analysis System, cameras record images of each field of view, which are then aligned to each other in order to determine which spots correspond to the same sequence template. The process is efficient but still requires enough spots to align images. Even in cases where samples have sufficient DNA to potentially generate millions of bases of sequences, template molecules could hybridize so sparsely that images cannot be aligned properly, resulting in poor sequencing results. In such cases, oligonucleotides of known sequence can be spiked into the samples at low quantity (typically at 40 pM) in order to provide a sufficiently high background level of spots to enable straightforward alignment of images, so that both the spiked and real samples can be read. The spiked sequences can then be filtered out from the resultant reads during the mapping procedure. A supplemental advantage of spiking is to provide a run-specific estimate of sequencing error rates by comparing the sequences of the reads to the known sequence of each spiked oligonucleotide.
In contrast to Helicos, standard Illumina sequencing is characterized by high reproducibility among lanes (Tables 1, 2). Furthermore, Illumina libraries could be reamplified and sequenced until exhaustion of the sequence complexity. No DNA amplification is performed in tSMS approaches, and overloaded and/or underloaded channels cannot be rerun. As most aDNA extraction methods are destructive, and as the source material is most often limited, projects that aim to achieve a complete draft of ancient genomes would be best served by using a combination of sequencing techniques and determining sample characteristics (DNA length and amounts, contamination, degree of modification) which are best suited to each technology (Table 2). That re-extracted undigested pellets (TC21a and TC21b) showed better performance than fresh extracts (TC21c) in recovering horse sequence information, suggests that optimizing extraction procedures, possibly focusing on putative preservation niches within the bones (Salamon et al. 2005), will be beneficial to all platforms.
The substantial levels of DNA damage that we identified toward the ends of both Illumina and Helicos sequence reads render alignment, and related SNP calling, challenging. The high rates of insertion and deletion (indel) sequencing errors that are specific to tSMS will complicate mapping even further, precluding accurate identification of indel variants unless large sequencing depth could be generated, which, in the case of ancient material, might be problematic and limited to a few exceptionally well-preserved specimens. Thus far, we have used both indexDP and BWA for aligning reads, because both are tolerant of indels (Li and Durbin 2009; Giladi et al. 2010). Mapping results showed good, albeit not complete overlap against the horse reference genome (∼70.0% of the reads uniquely mapped to the eqCab2 reference genome were found common to both aligners; data not shown). We therefore recommend using several mapping approaches when mapping aDNA sequence reads to reference genomes. Trimming sequence ends could improve mapping quality, as nucleotide misincorporations are mainly clustered at read termini. This conservative approach would result in loss of sequence information and would be more difficult with the short length of Helicos reads (here, median 31 bp). Illumina reads should be less problematic (here, median 67 bp), except in cases where aDNA templates have been highly fragmented. Integrating the nucleotide misincorporation and DNA fragmentation patterns in the mapping procedure would also improve mapping quality with less loss of sequence information. Indel-tolerant mapping softwares, like MIA (Mapping Iterative Assembler), which use position-specific scoring matrixes to estimate the mapping quality, could be advantageously used for that purpose. For now, we provide a preliminary estimate of the fraction of the endogenous reads that do not map against eqCab2 as a result of nucleotide misincorporation (Supplemental Text). Our estimate suggests that endogenous sequence yields reported here underestimate the overall Helicos performance, as 5.0% of the endogenous reads would not be mapped at all as long as one of the first two nucleotides of the reads corresponded to one deaminated Cytosine. The mapping of Illumina reads was found to be less sensitive to nucleotide misincorporation (0.8%), because of the longer sequence length.
In summary, we report the first in-depth analysis of tSMS of aDNA templates using the Helicos HeliScope Sequencer. We characterize the respective pros and cons in comparison with Illumina's GAIIx platform (Table 2). We show that a substantial fraction of the aDNA templates harbor 3′-overhanging termini, which are degraded during library preparation, and, hence, remain inaccessible to second-generation sequencing approaches. A series of other third-generation platforms are currently under development (Shendure and Ji 2008; Eid et al. 2009), but these approaches are likely to be challenged by the very short fragment sizes and low DNA concentrations found in aDNA. The data presented here is the first step toward using a combination of new sequencing tools tailored to individual sample properties, which will allow sequencing of paleogenomes in an unprecedented manner.
Methods
aDNA extraction and amplification
The horse fossil bone analyzed (TC21) originates from Pleistocene permafrost deposits at Thistle Creek, Yukon, and is associated with an infinite radiocarbon date (OxA-23933 > 50,300BP; UBA-16493 and UBA-17013 > 50,505BP). This fossil bone was extracted in aDNA facilities at the Center for GeoGenetics, using a combination of two silica-based methods. Briefly, a 3.6-g piece of bone was crushed to fine powder using a microdismembrator and first digested for 24 h at 55°C in 30 mL of 0.5 M EDTA. The undigested pellets were recovered the next day by spinning at 4000 rpm for 5 min and stored at −20°C, while the supernatant was further concentrated down to 250 μL using two 30-kDa Amicon centrifugal filter units (Millipore) and purified in 60 μL (30 μL from each of the two Amicon filters) of elution buffer. The data resulting from shotgun sequencing of this first extract are not presented in this study, but the remaining undigested pellets (UP) and 1.8 g of fine powder drilled at low speed from the same bone specimen (sample TC21c) were further extracted using a 48-h digestion in 5 and 7.5 ml of extraction buffer, respectively (0.5 M EDTA, 0.5% N-lauryl-Sarcosyl, 1 mg/mL Proteinase K at pH 8.0). The supernatant of TC21c and UP, recovered after spinning the solution at 2000 rpm for 2 min, were respectively transferred into 20 and 30 ml of binding solution with 100 and 200 μL of a fresh silica suspension prepared as described in Rohland and Hofreiter (2007). The final pH was adjusted to 4.0–5.0 using pH paper. DNA binding to silica surfaces was performed for 3 h at 37°C with agitation. After incubation, the volume of the UP solution was split into two equal parts (referred to as TC21a and TC21b). Silica particles were retrieved through spinning for 2 min at 12,000 rpm, washed twice with 80% ethanol before being eluted in 90 μL of elution buffer (QIAGEN), and stored at +4°C. The presence of DNA in the bone extracts was checked on a high-sensitivity lab-chip (Agilent; Supplemental Fig. 12) before being aliquoted and prepared either for second-generation sequencing or shipped to the Helicos BioSciences Corporation facilities for tSMS. In addition, horse-specific PCR amplification products of a 72-bp long mtDNA fragment were recovered (forward primer 5′-GATTTCCCGCGGCTTGGT; Reverse 5′-TCATTTCCAGYCAACA), suggesting: (1) that no DNA polymerase inhibitor that could have interfered with downstream library building and template preparation protocols was present in the extract, and (2) that the extract was not comprised solely of microbial environmental metagenomes, but contained a sufficient number of endogenous horse DNA fragments for further processing.
Illumina sequencing
DNA libraries were built in aDNA laboratory facilities in order to limit possible contamination issues. A DNA library was created as described in Meyer and Kircher (2010) without DNA fragmentation. A total of 15 μL of DNA extract (TC21c) was incubated for 15 min at 25°C, followed by 5 min at 12°C in buffer Tango supplemented with deoxynucleotide (final concentration: 100 mM), ATP (final concentration: 1 mM), and 35 U of T4 polynucleotide kinase and 7 U of T4 DNA polymerase. This step generated the 5′-phosphorylated blunt ends required for subsequent adapter ligation. DNA was purified using the MinElute PCR purification kit (QIAGEN) using 10 μL as elution volume. P5 and P7 adaptors (PE adaptor oligo mix) were further ligated by incubating the DNA eluate for 30 min at 22°C with an equal volume of a master mix consisting of a 2× T4 ligase buffer, 10% PEG-4000, 5 U of T4 ligase, and 5 mM of each adapter mix. DNA was purified (MinElute PCR purification kit; QIAGEN), and the adapter fill-in reaction was performed for 20 min at 37°C in a Thermopol buffer supplemented with 250 mM of each dNTP and 12 U of Bst Polymerase.
After a last column purification, the whole 10 μL of the DNA library was PCR amplified using a 50-μL reaction volume under the following conditions: 2.5 mM MgCl2, 1× TaqGold buffer, 0.2 μM each primer (5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTT, and 5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACC), 0.2 mM each dNTP, and 2 U of TaqGold. Cycling conditions consisted of an initial denaturation at 95°C for 9 min, followed by 24 cycles of denaturation at 95°C for 15 sec, annealing at 60°C for 20 sec, and extension at 72°C for 30 sec. A final extension was performed for 10 min at 72°C. A further reamplification under identical conditions was done for 10 cycles, except that 5 μL of the previous PCR was used as template for a total of 10 reactions. The quality of the library was further checked on a 2% agarose gel and DNA fragments ranging from ∼130 to 250 bp were gel-purified using the E.Z.N.A. gel-purification kit (Omega Bio-Tek). Overall, a total of five library amplifications were gel purified through one column and eluted with 30 μL of elution buffer (10 mM Tris-HCl at pH 8.5) prior to sequencing.
DNA sequencing was performed on the Illumina Genome Analyzer IIx platform available at the National High-throughput DNA Sequencing Center (Denmark) using seven lanes of 76 cycles on a single-read flow cell according to the manufacturer's instructions. The images were converted into intensity files and the Illumina base-calling pipeline (RTA1.8/SCS2.8) was run in order to generate fastq sequence files. Raw reads were further filtered to remove reads with bases determined as “N”, trimmed for residual adapter sequence, and regions starting or ending with a phred quality score of 2 using a program called SinglePrimerEndRemoval written in C++ (Stinus Lindgren, pers. comm.).
Helicos sequencing
Helicos HeliScope sequencing reactions were performed at the Helicos BioSciences Corporation facilities. A volume of 8 μL of DNA extracts was mixed with 2.8 μL of nuclease-free water, 2 μL of NEB Terminal Transferase 10× buffer, and 2 μL of a 2.5-mM CoCl2 solution, and heated at 80°C or 95°C for 5 min in a thermocycler for denaturation. Rapid cooling on ice was performed in order to minimize reannealing of denatured DNA strands. Single-stranded DNA molecules were poly(A) tailed for 1 h at 37°C. For the poly(A)-tailing reaction, the volume of the previous mix was increased to 20 μL through the addition of 5 U of NEB Terminal Transferase, NEB BSA (to 1 final concentration), and dATP at a final concentration of 10 μM in the 20-μL reaction. Reactions were stopped by inactivating the enzyme at 70°C for 10 min. As the DNA is prone to reannealing during the tailing step, heating at 80°C or 95°C, followed by rapid cooling, was repeated before 10 μL of 3′-end blocking master mix (NEB Terminal Transferase buffer, 250 μM CoCl2, 5 U of NEB Terminal Transferase, 10 μM of Biotin-ddATP) was added to the tailing reaction volume. The 3′-end blocking reactions were performed for 1 h at 37°C and stopped by denaturing the enzyme at 70°C for 20 min. DNA that may have reannealed during the blocking reaction was converted back to single strands by repeating the previous heating–rapid-cooling conditions prior to loading the samples on the flow cell. After addition of 10 μL of 2× hybridization buffer, 20 μL of sample was added to each channel and allowed to hybridize for 1 h at 37°C. The buffer was then rinsed away and the extra bases of the poly(A) tailed filled in with TTP and then locked in place with the first non-TTP base (Lipson et al. 2009). Sequencing was carried out using Virtual Terminator nucleotides as described in Bowers et al. (2009). The resulting sequence reads were then filtered for length (discarding sequences shorter than 25 nt) and for artifactual sequences that were too similar to the order of nucleotide addition (CTAG). When the sequence started with more than two T's, the leading T's were removed in case they arose from an incomplete fill and lock reaction. The remaining set of filtered reads was then analyzed.
DNA sequence analyses
The DNA sequence data analyzed in this study are available on NCBI Sequence Read Archive (SRA accession no. SRP005902, for both Illumina and Helicos reads). Filtered Illumina and Helicos reads were mapped against the horse and donkey mitogenomes ( accession no. NC_001640 and NC_001788, respectively), the horse reference genome (equCab2, filtered for the mitochondrial genome and chromosome Un), and the human reference genome (hg19) available for download at the UCSC Genome Bioinformatics website (http://genome.ucsc.edu/). Mitogenomes and nuclear genomes were mapped separately in order to avoid possible numt misidentification. Global alignments were performed with BWA (Li and Durbin 2009) after indexing the reference (mito)genomes using the index command and a linear-time algorithm. The Suffix Array coordinates of the reads showing a minimal size of 25 nt were found using the aln command and default parameters. The output was further converted in sam format with the samse command and reads mapping uniquely the horse reference genome, but not the human reference, regardless of the number of mismatches, were filtered for mapping quality scores higher than 25 using the samtools view command. This very conservative approach was performed in order to remove possible remnant human contamination and paralogs, as both could bias the analyses of DNA substitution and fragmentation patterns. Illumina reads starting and ending at the same coordinates were collapsed using the samtools rmdup command that keeps the read showing the highest mapping quality, as they could result from clonal expansion during library amplification. Finally, 1,000,000 random reads per lane (GAIIx) or channel (HeliScope) were analyzed using MegaBlast against the nucleotide database with a word size of 16, a gap opening penalty of −2, an identity percentage cut-off of 0.9, a maximal expect value 0.01, and default parameters otherwise in order to characterize the taxonomic origin of sequence reads. The megablast outputs were further assigned to major taxonomic groups using MEGAN 3.9 (Huson et al. 2007).
DNA fragmentation and misincorporation patterns were generated using the custom-made mapDamage package (Ginolhac et al. 2011), parsing quality filtered sam files as input, and recovering corresponding regions in reference genomes with samtools. mapDamage generated chromosome-specific output files reporting the frequencies of all possible substitutions and indels as a function of distance for 5′- to 3′-ends and 3′ to 5′ as well as read base composition. Furthermore, the base composition of the genomic regions located upstream and downstream (20 nt) of the reads was recorded. Statistical tests and misincorporation and fragmentation patterns were generated using custom R scripts (R Development Core Team 2010). The same patterns were analyzed using modern human DNA reads that are publicly available (Sequence Read Archive ID: SRA009216) in order to monitor for possible method specific substitution and base composition biases in sequencing. One of the eight fastq files consisting of 343,743,622 reads was downloaded and 3,000,000 randomly selected reads were mapped against hg19 and filtered for minimal quality scores of 25, resulting in 1,090,673 unique hits that were further analyzed using the mapDamage package. The mapDamage package is freely available with documentation and example files at http://geogenetics.ku.dk/all_literature/mapdamage/.
Data access
The sequence data generated in this study have been submitted to the NCBI Sequence Read Archive (http://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) under accession number SRP005902.
Acknowledgments
We thank Tina Brand, Jesper Stenderup, and the laboratory technicians at the Danish High-throughput DNA Sequencing Centre for technical assistance; Anders Krogh and Thomas Sicheritz-Ponten for access to computation facilities; Stuart Schmidt for assistance and support for the recovery of this and other Pleistocene fossils at Thistle Creek; Stinus Lindgreen, Mikkel Schubert, Anders Hansen, and Enrico Cappellini for fruitful discussions related to aDNA damage. This work was supported by the Danish Council for Independent Research, Natural Sciences (FNU); the Danish National Research Foundation; National Science Foundation ARC-0909456; the Searle Scholars Program; and the King Saud University Distinguished Scientist Fellowship Program (DSFP).
References
- ↵J Binladen, C Wiuf, MTP Gilbert, M Bunce, R Barnett, G Larson, AD Greenwood, J Haile, SYW Ho, AJ Hansen, . 2006. Assessing the fidelity of ancient DNA sequences amplified from nuclear genes. Genetics 172: 733–741.
- ↵J Bowers, J Mitchell, E Beer, PR Buzby, M Causey, JW Efcavitch, M Jarosz, E Krzymanska-Olejnik, L Kung, D Lipson, . 2009. Virtual terminator nucleotides for next-generation DNA sequencing. Nat Methods 6: 593–595.
- ↵B Bramanti, MG Thomas, W Haak, M Unterlaender, P Jores, K Tambets, I Antanaitis-Jacobs, MN Haidle, R Jankauskas, C-J Kind, . 2009. Genetic discontinuity between local hunter-gatherers and Europe's first farmers. Science 326: 137–140.
- ↵AW Briggs, U Stenzel, PLF Johnson, RE Green, J Kelso, K Prufer, M Meyer, J Krause, MT Ronan, M Lachmann, . 2007. Patterns of damage in genomic DNA sequences from a Neandertal. Proc Natl Acad Sci 104: 14616–14621.
- ↵AW Briggs, JM Good, RE Green, J Krause, T Maricic, U Stenzel, C Lalueza-Fox, P Rudan, D Brajkovic, Z Kucan, . 2009. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science 325: 318–321.
- ↵P Brotherton, P Endicott, JJ Sanchez, M Beaumont, R Barnett, J Austin, A Cooper. 2007. Novel high-resolution characterization of ancient DNA reveals C > U-type base modification events as the sole cause of post mortem miscoding lesions. Nucleic Acids Res 35: 5717–5728.
- ↵HA Burbano, E Hodges, RE Green, AW Briggs, J Krause, M Meyer, JM Good, T Maricic, PLF Johnson, Z Xuan, . 2010. Targeted investigation of the Neandertal genome by array-based sequence capture. Science 328: 723–725.
- ↵PF Campos, E Willerslev, A Sher, L Orlando, E Axelsson, A Tikhonov, K Aaris-Sørensen, AD Greenwood, R-D Kahlke, P Kosintsev, . 2010. Ancient DNA analyses exclude humans as the driving force behind late Pleistocene musk ox (Ovibos moschatus) population dynamics. Proc Natl Acad Sci 107: 5675–5680.
- ↵A Cooper, H Poinar. 2000. Ancient DNA: do it right or not at all. Science 289: 1139. doi: 10.1126/science.289.5482.1139b.
- ↵M de Bruyn, BL Hall, LF Chauke, C Baroni, PL Koch, AR Hoelzel. 2009. Rapid response of a marine mammal species to Holocene climate and habitat change. PLoS Genet 5: e1000554. doi: 10.1371/journal.pgen.1000554.
- ↵J Eid, A Fehr, J Gray, K Luong, J Lyle, G Otto, P Peluso, D Rank, P Baybayan, B Bettman, . 2009. Real-time DNA sequencing from single polymerase molecules. Science 323: 133–138.
- ↵E Giladi, J Healy, G Myers, C Hart, P Kapranov, D Lipson, S Roels, E Thayer, S Letovsky. 2010. Error tolerant indexing and alignment of short reads with covering template families. J Comput Biol 17: 1397–1411.
- ↵MTP Gilbert, AJ Hansen, E Willerslev, L Rudbeck, I Barnes, N Lynnerup, A Cooper. 2003. Characterization of genetic miscoding lesions caused by postmortem damage. Am J Hum Genet 72: 48–61.
- ↵MTP Gilbert, H-J Bandelt, M Hofreiter, I Barnes. 2005. Assessing ancient DNA. Trends Ecol Evol 20: 541–544.
- ↵MT Gilbert, J Binladen, W Miller, C Wiuf, E Willerslev, H Poinar, JE Carlson, JH Leebens-Mack, SC Schuster. 2007a. Recharacterization of ancient DNA miscoding lesions: insights in the era of sequencing-by-synthesis. Nucleic Acids Res 25: 1–10.
- ↵MTP Gilbert, LP Tomsho, S Rendulic, M Packard, DI Drautz, A Sher, A Tikhonov, L Dalén, T Kuznetsova, P Kosintsev, . 2007b. Whole-genome shotgun sequencing of mitochondrial from ancient hair shafts. Science 317: 1927–1930.
- ↵MTP Gilbert, T Kivisild, B Gronnow, PK Andersen, E Metspalu, M Reidla, E Tamm, E Axelsson, A Gotherstrom, PF Campos, . 2008. Paleo-eskimo mtDNA genome reveals matrilineal discontinuity in Greenland. Science 320: 1787–1789.
- ↵A Ginolhac, M Rasmussen, MTP Gilbert, E Willerslev, L Orlando. 2011. mapDamage: testing for damage patterns in ancient DNA sequences. Bioinformatics 27: 2153–2155.
- ↵RE Green, J Krause, SE Ptak, AW Briggs, MT Ronan, JF Simons, L Du, M Egholm, JM Rothberg, M Paunovic, . 2006. Analysis of one million base pairs of Neanderthal DNA. Nature 444: 330–336.
- ↵RE Green, A-S Malaspinas, J Krause, AW Briggs, PLF Johnson, C Uhler, M Meyer, JM Good, T Maricic, U Stenzel, . 2008. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell 134: 416–426.
- ↵RE Green, J Krause, AW Briggs, T Maricic, U Stenzel, M Kircher, N Patterson, H Li, W Zhat, MH-Y Fritz, . 2010. A draft sequence of the Neandertal genome. Science 328: 710–722.
- ↵W Haak, O Balanovsky, JJ Sanchez, S Koshel, V Zaporozhchenko, CJ Adler, CSI Der Sarkissian, G Brandt, C Schwarz, N Nicklisch, . 2010. Ancient DNA from European early Neolithic farmers reveals their near eastern affinities. PLoS Biol 8: e1000536. doi: 10.1371/journal.pbio.1000536.
- ↵E Hagelberg, B Sykes, R Hedges. 1989. Ancient bone DNA amplified. Nature 324: 485. doi: 10.1038/342485a0.
- ↵J Haile, DG Froese, RDE MacPhee, RG Roberts, LJ Arnold, AV Reyes, M Rasmussen, R Nielsen, BW Brook, S Robinson, . 2009. Ancient DNA reveals late survival of mammoth and horse in interior Alaska. Proc Natl Acad Sci 106: 22352–22357.
- ↵AJ Hansen, E Willerslev, C Wiuf, T Mourier, P Arctander. 2001. Statistical evidence for miscoding lesions in ancient DNA templates. Mol Biol Evol 18: 262–265.
- ↵TD Harris, PR Buzby, H Babcock, E Beer, J Bowers, I Braslavsky, M Causey, J Colonell, J DeMeo, JW Efcavitch, . 2008. Single-molecular DNA sequencing of a viral genome. Science 320: 106–109.
- ↵C Hart, D Lipson, F Ozsolak, T Raz, K Steinmann, J Thompson, PM Milos. 2010. Single-molecule sequencing: sequence methods to enable accurate quantitation. Methods Enzymol 472: 407–430.
- ↵R Higuchi, B Bowman, M Freiberger, OA Ryder, AC Wilson. 1984. DNA sequences from the quagga, an extinct member of the horse family. Nature 312: 282–284.
- ↵LW Hillier, GT Marth, AR Quinlan, D Dooling, G Fewell, D Barnett, P Fox, JI Glasscock, M Hickenbotham, W Huang, . 2008. Whole-genome sequencing and variant discovery in C. elegans. Nat Methods 5: 183–188.
- ↵SY Ho, TH Heupnik, A Rambaut, B Shapiro. 2007. Bayesian estimation of sequence damage in ancient DNA. Mol Biol Evol 24: 1416–1422.
- ↵M Hofreiter, V Jaenicke, D Serre, A Haeseler Av, S Paabo. 2001. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res 29: 4793–4799.
- ↵M Hoss, P Jaruga, TH Zasrawny, M Dizdaroglu, S Paabo. 1996. DNA damage and DNA sequence retrieval from ancient tissues. Nucleic Acids Res 24: 1304–1307.
- ↵D Huson, AF Auch, SC Schuster. 2007. MEGAN analysis of metagenomic data. Genome Res 17: 377–386.
- ↵DS Johnson, A Mortazavi, RM Myers, B Wold. 2007. Genome-wide mapping of in vivo protein-DNA interactions. Science 316: 1497–1502.
- ↵JA Kimbrel, SA Givan, AB Halgren, AL Creason, DI Mills, GM Banowetz, DJ Armstrong, JH Chang. 2010. An improved, high-quality draft genome sequence of the germination-arrest factor-producing Pseudomonas fluorescens WH6. BMC Genomics 11: 552. doi: 10.1186/1471-2164-11-522.
- ↵J Krause, PH Dear, JL Pollack, M Slatkin, H Spriggs, I Barnes, AM Lister, I Ebersberger, S Paabo, M Hofreiter. 2006. Multiplex amplification of the mammoth mitochondrial genome and the evolution of Elephantidae. Nature 439: 724–727.
- ↵J Krause, Q Fu, JM Good, B Viola, MV Shunkov, AP Derevianko, S Paabo. 2010a. The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature 464: 894–897.
- ↵J Krause, AW Briggs, M Kircher, T Maricic, N Zwyns, A Derevianko, S Paabo. 2010b. A complete mtDNA genome of an early modern human from Kostenki, Russia. Curr Biol 20: 231–236.
- ↵M Kuch, N Rohland, JL Betancourt, C Latorre, S Steppan, HN Poinar. 2002. Molecular analysis of a 11700-year-old rodent midden from the Atacama Desert, Chile. Mol Ecol 11: 913–924.
- ↵H Li, R Durbin. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760.
- ↵T Lindahl. 1993. Instability and decay of the primary structure of DNA. Nature 362: 709–715.
- ↵D Lipson, T Raz, A Kieu, DR Jones, E Giladi, E Thayer, JF Thompson, S Letovsky, P Milos, M Causey. 2009. Quantification of the yeast transcriptome by single-molecule sequencing. Nat Biotechnol 27: 652–658.
- ↵H Malmstrom, MTP Gilbert, MG Thomas, M Brandstrom, J Stora, P Molnar, PK Andersen, C Bendixen, G Holmlund, A Gotherstrom, . 2009. Ancient DNA reveals lack of continuity between Neolithic hunter-gatherers and contemporary Scandinavians. Curr Biol 19: 1758–1762.
- ↵T Maricic, M Whitten, S Paabo. 2010. Multiplexed SNA sequence capture of mitochondrial genomes using PCR products. PLoS ONE 5: e14004. doi: 10.1371/journal.pone.0014004.
- ↵M Metzker. 2010. Sequencing technologies—the next generation. Nat Rev Genet 11: 31–46.
- ↵M Meyer, M Kircher. 2010. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc doi: 10.1101/pdb.prot5448.
- ↵W Miller, DI Drautz, A Ratan, B Pusey, J Qi, AM Lesk, LP Tomsho, MD Packard, F Zhao, A Sher, . 2008. Sequencing the nuclear genome of the extinct woolly mammoth. Nature 456: 387–390.
- ↵D Mitchell, E Willerslev, A Hansen. 2005. Damage and repair of ancient DNA. Mutat Res 571: 265–276.
- ↵JP Noonan, M Hofreiter, D Smith, JR Priest, N Rohland, G Rabeder, J Krause, JC Detter, S Paabo, EM Rubin. 2005. Genomic sequencing of Pleistocene cave bears. Science 309: 597–599.
- ↵JP Noonan, G Coop, S Kudaravalli, D Smith, J Krause, J Alessi, F Chen, D Platt, S Paabo, JK Pritchard, . 2006. Sequencing and analysis of Neanderthal genomic DNA. Science 314: 1113–1118.
- ↵CL Oskam, J Haile, E McLay, P Rigby, ME Allentoft, ME Olsen, C Bengtsson, GH Miller, JL Schwenninger, C Jacomb, . 2010. Fossil avian eggshell preserves ancient DNA. Proc Biol Sci 277: 1991–2000.
- ↵S Paabo. 1989. Ancient DNA: extraction, characterization, molecular cloning, and enzymatic amplification. Proc Natl Acad Sci 86: 1939–1943.
- ↵S Paabo, RG Higuchi, AC Wilson. 1989. Ancient DNA and the polymerase chain reaction. J Biol Chem 264: 9709–9712.
- ↵S Paabo, H Poinar, D Serre, V Jaenicke-Despres, J Hebler, N Rohland, M Kuch, J Krause, L Vigilant, M Hofreiter. 2004. Genetic analyses from ancient DNA. Annu Rev Genet 38: 645–679.
- ↵H Poinar, M Kuch, G McDonald, P Martin, S Paabo. 2003. Nuclear gene sequences from a late pleistocene sloth coprolithe. Curr Biol 13: 1150–1152.
- ↵HN Poinar, C Schwarz, J Qi, B Shapiro, RDE MacPhee, B Buigues, A Tikhonov, DH Huson, LP Tomsho, A Auch, . 2006. Metagenomics to paleogenomics: Large-scale sequencing of mammoth DNA. Science 311: 392–394.
- ↵M Pruvost, R Schwarz, VB Correia, S Champlot, S Braguier, N Morel, Y Fernandez-Jalvo, T Grange, E-M Geigl. 2007. Freshly excavated fossil bones are best for amplification of ancient DNA. Proc Natl Acad Sci 104: 739–744.
- ↵D Pushkarev, NF Neff, SR Quake. 2009. Single-molecule sequencing of an individual human genome. Nat Biotechnol 27: 847–850.
- ↵MA Quail, I Kozarewa, F Smith, A Scally, PJ Stephens, R Durbin, H Swerdlow, DJ Turner. 2008. A large genome center's improvements to the Illumina sequencing system. Nat Methods 5: 1005–1010.
- ↵R Development Core Team. 2010. R: A language and envionment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org.
- ↵O Ramirez, E Gigli, P Bover, JA Alcover, J Bertranpetit, J Castresana, C Lalueza-Fox. 2009. Paleogenomics in a temperate environment: Shotgun sequencing from an extinct Mediterranean caprine. PLoS ONE 4: e5670. doi: 10.1371/journal.pone.0005670.
- ↵M Rasmussen, Y Li, S Lindgreen, JS Pedersen, A Albrechtsen, I Moltke, M Metspalu, E Metspalu, T Kivisild, R Gupta, . 2010. Ancient human genome sequence of an extinct paleo-eskimo. Nature 463: 757–762.
- ↵D Reich, RE Green, M Kircher, J Krause, N Patterson, EY Durand, B Viola, AW Briggs, U Stenzel, PLF Johnson, . 2010. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 468: 1053–1060.
- ↵N Rohland, M Hofreiter. 2007. Ancient DNA extraction from bones and teeth. Nat Protoc 2: 1756–1762.
- ↵N Rohland, AS Malaspinas, JL Pollack, M Slatkin, P Matheus, M Hofreiter. 2007. Proboscidean mitogenomics: chronology and mode of elephant evolution using mastodon as outgroup. PLoS Biol 5: e207. doi: 10.1371/journal.pbio.0050207.
- ↵N Rohland, D Reich, S Mallick, M Meyer, RE Green, NJ Georgiadis, AL Roca, M Hofreiter. 2010. Genomic DNA sequences from mastodon and woolly mammoth reveal deep speciation of forest and savanna elephants. PLoS Biol 8: e1000564. doi: 10.1371/journal.pbio.1000564.
- ↵H Rompler, N Rohland, C Lalueza-Fox, E Willerslev, T Kuznetsova, G Rabeder, J Bertranpetit, T Schoneberg, M Hofreiter. 2006. Nuclear gene indicates coat-color polymorphism in mammoths. Science 313: 62. doi: 10.1126/science.1128994.
- ↵RK Saiki, S Scharf, F Faloona, KB Mullis, GT Horn, HA Erlich, N Arnheim. 1985. Enzymatic amplification of β-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230: 1350–1354.
- ↵M Salamon, N Tuross, B Arensburg, S Weiner. 2005. Relatively well preserved DNA is present in the crystal aggregates of fossil bones. Proc Natl Acad Sci 102: 13783–13788.
- ↵C Schwarz, R Debruyne, M Kuch, E McNally, H Schwarcz, AD Aubrey, J Bada, H Poinar. 2009. New insights from old bones: DNA preservation and degradation in permafrost preserved mammoth remains. Nucleic Acids Res 37: 3215–3229.
- ↵B Shapiro, AJ Drummond, A Rambaut, MC Wilson, PE Matheus, AV Sher, OG Pybus, MTP Gilbert, I Barnes, J Binladen, . 2004. Rise and fall of the Beringian steppe bison. Science 306: 1561–1565.
- ↵J Shendure, H Ji. 2008. Next-generation DNA sequencing. Nat Biotechnol 26: 1135–1145.
- ↵M Stiller, RE Green, M Ronan, JF Simons, L Du, W He, M Egholm, JM Rothberg, SG Keates, ND Ovodov, . 2006. Patterns of nucleotide misincorporations during enzymatic amplification and direct large-scale sequencing of ancient DNA. Proc Natl Acad Sci 103: 13578–13584.
- ↵M Stiller, G Baryshnikov, H Bocherens, AG D'Anglade, B Hilpert, SC Munzel, R Pinhasi, G Rabeder, W Rosendahl, E Trinkaus, . 2010. Withering away– 25,000 years of genetic decline preceded cave bear extinction. Mol Biol Evol 27: 975–978.
- ↵JF Thompson, KE Steinmann. 2010. Single molecular sequencing with a HeliScope genetic analysis system. Curr Protoc Mol Biol 7: doi: 10.1002/0471142727.mb0710s92.
- ↵SK Wall, JD Kim. 2007. Inconsistencies in Neanderthal genomic DNA sequences. PLoS Genet 3: e175. doi: 10.1371/journal.pgen.0030175.
- ↵E Willerslev, A Cooper. 2005. Ancient DNA. Proc Biol Sci 272: 3–16.
- ↵E Willerslev, AJ Hansen, J Binladen, TB Brand, MTP Gilbert, B Shapiro, M Bunce, C Wiuf, DA Gilichinsky, A Cooper. 2003. Diverse plant and animal genetic records from Holocene and Pleistocene sediments. Science 300: 791–795.
- ↵E Willerslev, AJ Hansen, R Ronn, TB Brand, I Barnes, C Wiuf, D Gilichinsky, D Mitchell, A Cooper. 2004. Long-term persistence of bacterial DNA. Curr Biol 14: R9–R10.
- ↵E Willerslev, E Cappellini, W Boomsma, R Nielsen, MB Hebsgaard, TB Brand, M Hofreiter, M Bunce, HN Poinar, D Dahl-Jensen, . 2007. Ancient biomolecules from deep ice cores reveal a forested southern Greenland. Science 317: 111–114.
- ↵E Willerslev, MTP Gilbert, J Binladen, SYW Ho, PF Campos, A Ratan, LP Tomsho, RR da Fonseca, A Sher, TV Kuznetsova, . 2009. Analysis of complete mitochondrial genomes from extinct rhinoceroses reveal lack of phylogenetic resolution. BMC Evol Biol 9: 95. doi: 10.1186/1471-2148-9-95.
Notes
[1] [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.122747.111.