
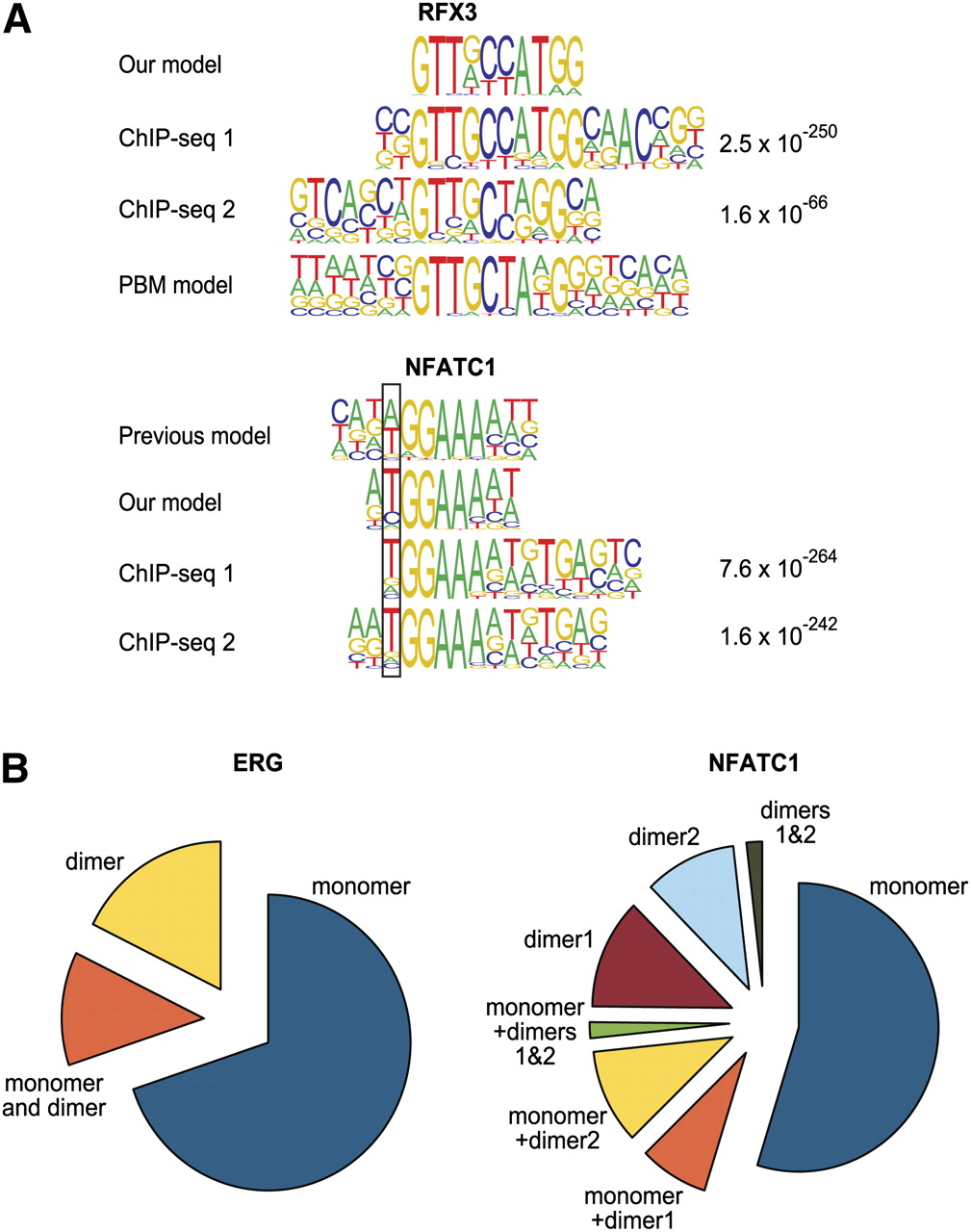
Validation of binding models by ChIP-seq. (A) Comparison of in vitro derived binding models for RFX3 and NFATC1 and previously described models (Kel et al. 1999; Badis et al. 2009) with motifs that are enriched in peaks from a ChIP-seq experiment for the same factors. The MEME algorithm was used to identify enriched motifs in the ChIP-seq peaks. For both factors, two different antibodies were used in the experiments shown; the expectation value for the motifs is indicated on the right. (Top) Note that both our model and the model of Badis et al. (2009) (PBM model) are supported by ChIP-seq. For NFATC1, the position where our model matches the ChIP-seq-derived model better than the previous model is boxed. Note also that the ChIP-seq-enriched motif for NFATC1 (bottom) appears to contain also signal (g/a)TGA(g/c) that is located right of the NFATC1 monomer profile tGGAAAa(t/a). This signal is likely derived from a dimerization partner, as it is well known that NFAT proteins dimerize with many other TFs (Macian 2005). (B) Relative fractions of peaks containing monomer and dimer sites and combinations thereof for NFATC1 and ERG (ChIP-seq data from Wei et al. 2010). For all matrices, the cut-off score was set to yield 1 site per 10 kb of human genome. Note that a significant fraction of peaks contain motifs that match the dimer model but not the monomer model. The total fraction of peaks with sites was 24% and 14% for NFATC1 and ERG, respectively.








