
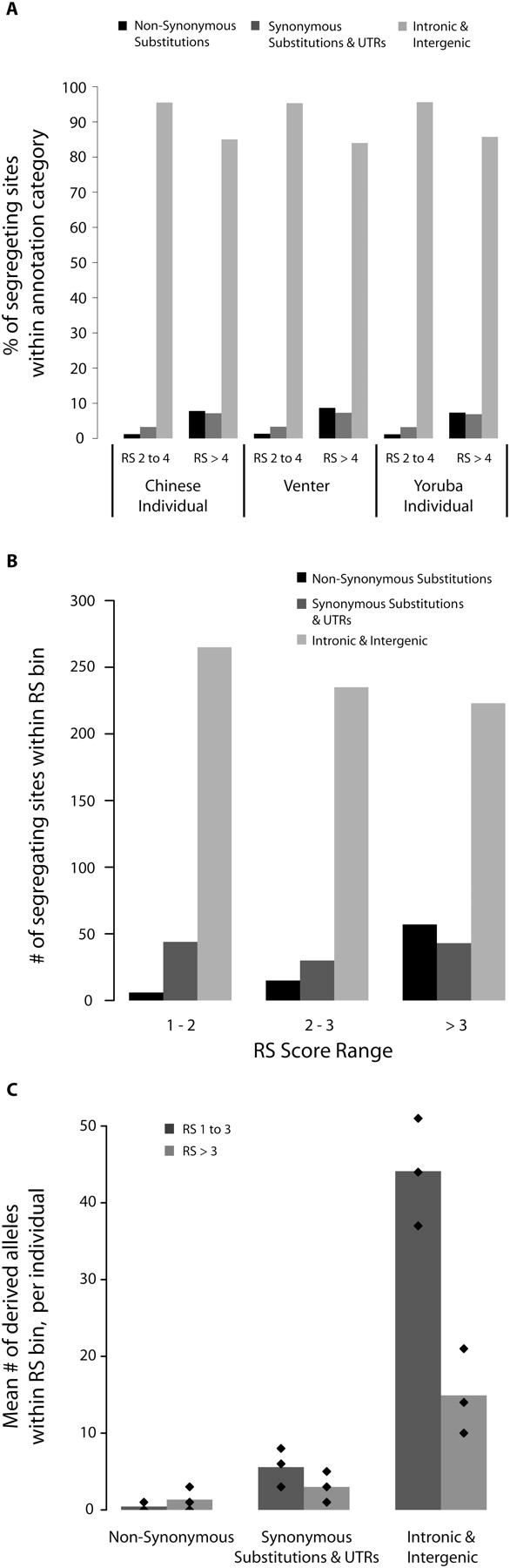
Relative abundance of coding and noncoding functional variation. SNVs were divided into three categories: those that cause nonsynonymous substitutions; those that cause synonymous substitutions or changes in the UTRs; and those that do not occur in exons (intronic and intergenic). (A) Contribution of SNVs in each category to total variation at constrained sites in each resequenced individual genome at sites with RS 2 to 4 and at sites with RS > 4. (B) Total number of segregating sites in our ENCODE resequencing sample that occur at constrained sites in our three annotation categories. Constrained sites are divided into bins of increasing constraint. (C) Mean number of segregating sites carried by the individuals in our ENCODE resequencing sample in all three categories, at moderately (RS 1 to 3) and highly (RS > 3) constrained positions. Diamonds correspond to the 10%, 50%, and 90% quantiles.








