
Abstract
Next-generation massively parallel DNA sequencing technologies provide ultrahigh throughput at a substantially lower unit data cost; however, the data are very short read length sequences, making de novo assembly extremely challenging. Here, we describe a novel method for de novo assembly of large genomes from short read sequences. We successfully assembled both the Asian and African human genome sequences, achieving an N50 contig size of 7.4 and 5.9 kilobases (kb) and scaffold of 446.3 and 61.9 kb, respectively. The development of this de novo short read assembly method creates new opportunities for building reference sequences and carrying out accurate analyses of unexplored genomes in a cost-effective way.
The development and commercialization of next-generation massively parallel DNA sequencing technologies, including Illumina Genome Analyzer (GA) (Bentley 2006), Applied Biosystems SOLiD System, and Helicos BioSciences HeliScope (Harris et al. 2008), have revolutionized genomic research. Compared to traditional Sanger capillary-based electrophoresis systems, these new technologies provide ultrahigh throughput with two orders of magnitude lower unit data cost. However, they all share a common intrinsic characteristic of providing very short read length, currently 25–75 base pairs (bp), which is substantially shorter than the Sanger sequencing reads (500–1000 bp) (Shendure et al. 2004). This has raised concern about their ability to accurately assemble large genomes. Illumina GA technology has been shown to be feasible for use in human whole-genome resequencing and can be used to identify single nucleotide polymorphisms (SNPs) accurately by mapping the short reads onto the known reference genome (Bentley et al. 2008; Wang et al. 2008). But to thoroughly annotate insertions, deletions, and structural variations, de novo assembly of each individual genome from these raw short reads is required.
Currently, Sanger sequencing technology remains the dominant method for building a reference genome sequence for a species. It is, however, expensive, and this prevents many genome sequencing projects from being put into practice. Over the past 10 yr, only a limited number of plant and animal genomes have been completely sequenced, (http://www.ncbi.nlm.nih.gov/genomes/static/gpstat.html), including human (Lander et al. 2001; Venter et al. 2001) and mouse (Mouse Genome Sequencing Consortium 2002), but accurate understanding of evolutionary history and biological processes at a nucleotide level requires substantially more. The development of a de novo short read assembly method would allow the building of reference sequences for these unexplored genomes in a very cost-effective way, opening the door for carrying out numerous substantial new analyses.
Several programs, such as phrap (http://www.phrap.org), Celera assembler (Myers et al. 2000), ARACHNE (Batzoglou et al. 2002), Phusion (Mullikin and Ning 2003), RePS (Wang et al. 2002), PCAP (Huang et al. 2003), and Atlas (Havlak et al. 2004), have been successfully used for de novo assembly of whole-genome shotgun (WGS) sequencing reads in the projects applying the Sanger technology. These are based on an overlap-layout strategy, but for very short reads, this approach is unsuitable because it is hard to distinguish correct assembly from repetitive sequence overlap due to there being only a very short sequence overlap between these short reads. Also, in practice, it is unrealistic to record into a computer memory all the sequence overlap information from deep sequencing that are made up of huge numbers of short reads.
The de Bruijn graph data structure, introduced in the EULER (Pevzner et al. 2001) assembler, is particularly suitable for representing the short read overlap relationship. The advantage of the data structure is that it uses K-mer as vertex, and read path along the K-mers as edges on the graph. Hence, the graph size is determined by the genome size and repeat content of the sequenced sample, and in principle, will not be affected by the high redundancy of deep read coverage. A few short read assemblers, including Velvet (Zerbino and Birney 2008), ALLPATHS (Butler et al. 2008), and EULER-SR (Chaisson and Pevzner 2008), have adopted this algorithm, explicitly or implicitly, and have been implemented and shown very promising performances. Some other short read assemblers have applied the overlap and extension strategy, such as SSAKE (Warren et al. 2007), VCAKE (Jeck et al. 2007) (the follower of SSAKE which can handle sequencing errors), SHARCGS (Dohm et al. 2007), and Edena (Hernandez et al. 2008). However, all these assemblers were designed to handle bacteria- or fungi-sized genomes, and cannot be applied for assembly of large genomes, such as the human, given the limits of the available memory of current supercomputers. Recently, ABySS (Simpson et al. 2009) used a distributed de Bruijn graph algorithm that can split data and parallelize the job on a Linux cluster with message passing interface (MPI) protocol, allowing communication between nodes. Thus, it is able to handle a whole short read data set of a human individual; however, the assembly is very fragmented with an N50 length of ∼1.5 kilobases (kb). This is not long enough for structural variation detection between human individuals, nor is it good enough for gene annotation and further analysis of the genomes of novel species.
Here, we present a novel short read assembly method that can build a de novo draft assembly for the human genome. We previously sequenced the complete genome of an Asian individual using a resequencing method, producing a total of 117.7 gigabytes (Gb) of data, and have now an additional 82.5 Gb of paired-end short reads, achieving a 71× sequencing depth of the NCBI human reference sequence. We used this substantial amount of data to test our de novo assembly method, as well as the data from the African genome sequence (Bentley et al. 2008; Wang et al. 2008; Li et al. 2009a). We compared the de novo assemblies to the NCBI reference genome and demonstrated the capability of this method to accurately identify structural variations, especially small deletions and insertions that are difficult to detect using the resequencing method. This software has been integrated into the short oligonucleotide alignment program (SOAP) (Li et al. 2008, 2009b,c) package and named SOAPdenovo to indicate its functionality.
Results
Genome repeat structure and predicted assembly
The main difficulty of assembling a shotgun short read data set into a complete genome is the presence of repetitive sequences that have multiple identical or very similar copies in the genome. Thus, analyzing the repeat structure of a known reference genome or closely related species would help for designing the sequencing project and provide a theoretical estimation of the expected assembly.
In humans, about half of the genome is derived from transposable elements (TEs) (Lander et al. 2001). Most transposons are under neutral selection, so new copies will accumulate mutations quickly after duplication and will become easily distinguishable from the other repeat copies. On analyzing the human genome, we found that ∼79% of the sequence was composed of unique 25-mers. Length distribution of the continuous repetitive 25-mers showed that over 47% of the repeat clusters are shorter than 1 kb (Fig. 1A). There are two peaks with repeat-cluster lengths of about 300 bp and 6 kb, which correspond to the two most abundant TE classes in the human genome: Alu and L1 retrotransposons, respectively. Over 78% of the unique clusters range between 500 bp and 5 kb. So, theoretically, at an appropriate sequencing depth, using a 25-mer as the node size for assembly, the expected contig N50 size of the unique sequences will be 1.3 kb; reducing seed size to a 21-mer, the expected contig N50 size would be as short as 251 bp; and increasing it to a 29-mer gives an expected contig N50 of 1.9 kb (Supplemental Fig. 1). Bigger K-mers would give longer contig sizes, but would require deeper sequencing or longer read length to guarantee that short reads overlap more than a selected K-mer size at each genomic location.
(A) Length distribution of unique and repeat sequence clusters in the human genome. At each chromosomal location, we checked the frequency of the 25-mer in the whole human genome. If it appeared once, we defined it as unique; otherwise it was considered a repeat 25-mer. The regions were then merged as unique clusters and repeat clusters, and those small unique clusters (<100 bp) inside repeat clusters were defined as repeats. (B) Sequence length distribution of an ideal assembly with each insert-sized paired-ends. The repeat clusters with lengths smaller than the assumed insert size of paired-ends were crossed and the unique clusters were merged. These unique clusters represent the ideal assembly using the paired-ends.
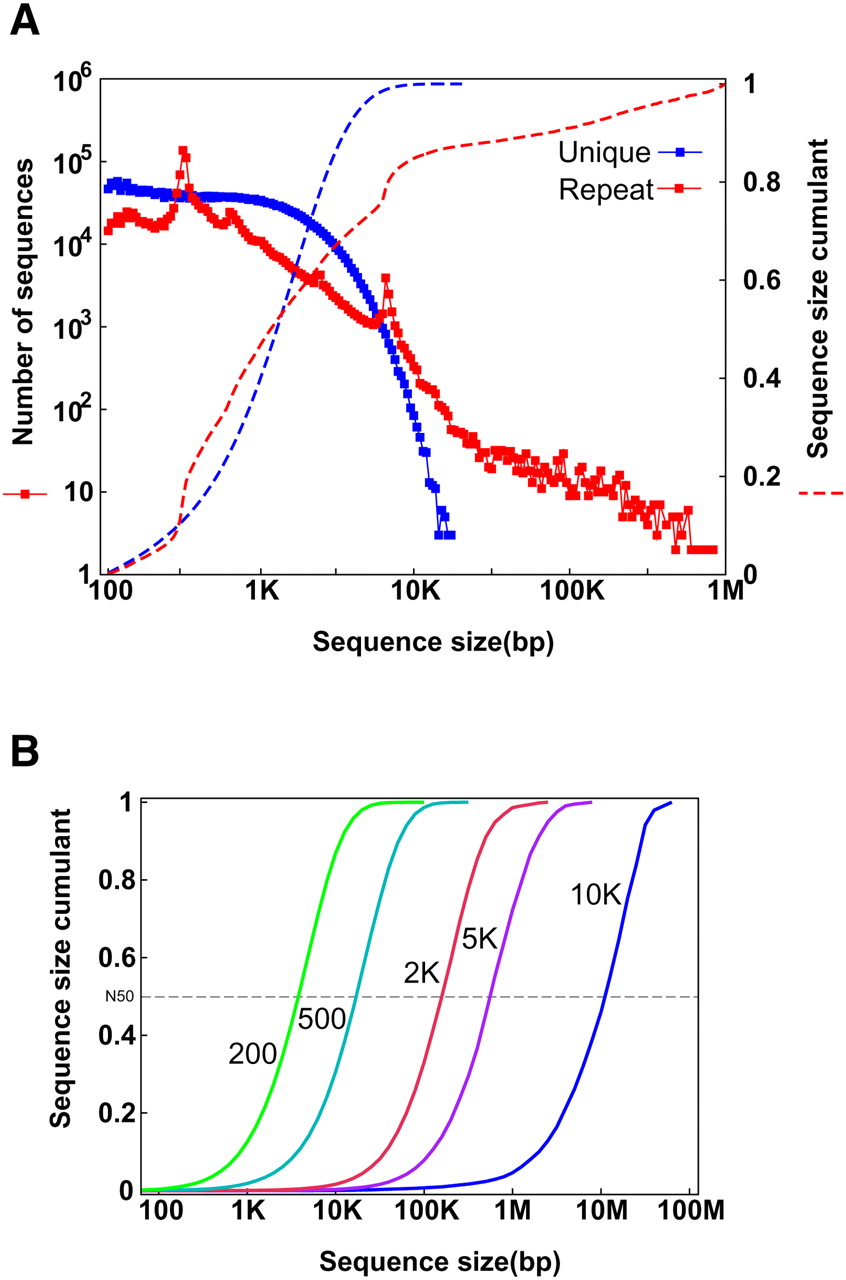
Resolving the repeat clusters between unique clusters and assembling them into an intact sequence requires the paired-end relationship of a pair of short reads generated from both ends of a DNA clone. If both of the two reads are unique and located on two neighboring unique clusters, then we can order these two unique clusters, estimate the distance according to the clone insert size, fill in the internal repeat cluster, and join them into a long sequence. Thus, in principle, a repeat cluster of size N could be crossed by paired-ends with a clone insert size longer than N. Using 200, 500, 2000, 5000, and 10,000 bp insert size of paired-ends, the expected scaffold N50 size of the human genome is 4, 18, 158, 562, and 9870 kb, respectively (Fig. 1B). Of course, to fill in the intrascaffold gaps effectively, and avoid interleaving, stepwise paired-end insert sizes would be needed.
Overall strategy for large genome assembly
We sequenced 200 Gb of Illumina GA reads for the Asian individual, including 72-Gb single-end and 128-Gb paired-end reads. The read lengths ranged from 35 bp to 75 bp, and the insert sizes of the paired-end libraries were 140 bp, 440 bp, 2.6 kb, 6 kb, and 9.6 kb (Supplemental Table 1). To manage the huge number of short reads effectively and handle them in a standard supercomputer with 512 Gb memory installed, we modularized the assembly method and organized it as a pipeline by loading only the necessary data at each step. Since some (∼5%) chimeric reads in long paired-end (≥2 kb) sequencing are generated in the circularizing and fragmentation process (Bentley et al. 2008), we only used single-end and paired-end reads with insert sizes of 140 bp and 440 bp for contig assembly; all paired-end data were used for scaffold construction.
We used genomic DNA to construct sequencing libraries and generated short reads from both ends of the clones (Fig. 2A). The read sequences were loaded into the computer and de Bruijn graph data structure was used to represent the overlap among the reads (Fig. 2B). Next, erroneous connections in the graph were removed, tiny repeats were resolved by read path, and the graph was simplified by merging unambiguously connected nodes into one (Fig. 2C). There are three major types of erroneous connections that need to be addressed: (1) tips—short and low-coverage dead ends, which are likely to be caused by sequencing errors at read ends; (2) low-coverage links—nodes connected by only one or a few reads, which are likely to be chimeric connections; and (3) bubbles—redundant paths with minor differences, which may represent polymorphisms between either homogenous chromosomes or repeat copies.
Schematic overview of the assembly algorithm. (A) Genomic DNA was fragmented randomly and sequenced using paired-end technology. Short clones with sizes between 150 and 500 bp were amplified and sequenced directly; while long range (2–10 kb) paired-end libraries were constructed by circularizing DNA, fragmentation, and then purifying fragments with sizes in the range of 400–600 bp for cluster formation. (B) The raw or precorrected reads were then loaded into computer memory and de Bruijn graph data structure was used to represent the overlap among the reads. (C) The graph was simplified by removing erroneous connections (in red color on the graph) and solving tiny repeats by read path: (i) Clipping the short tips, (ii) removing low-coverage links, (iii) solving tiny repeats by read path, and (iv) merging the bubbles that were caused by repeats or heterozygotes of diploid chromosomes. (D) On the simplified graph, we broke the connections at repeat boundaries and output the unambiguous sequence fragments as contigs. (E) We realigned the reads onto the contigs and used the paired-end information to join the unique contigs into scaffolds. (F) Finally, we filled in the intrascaffold gaps, which were most likely comprised by repeats, using the paired-end extracted reads.
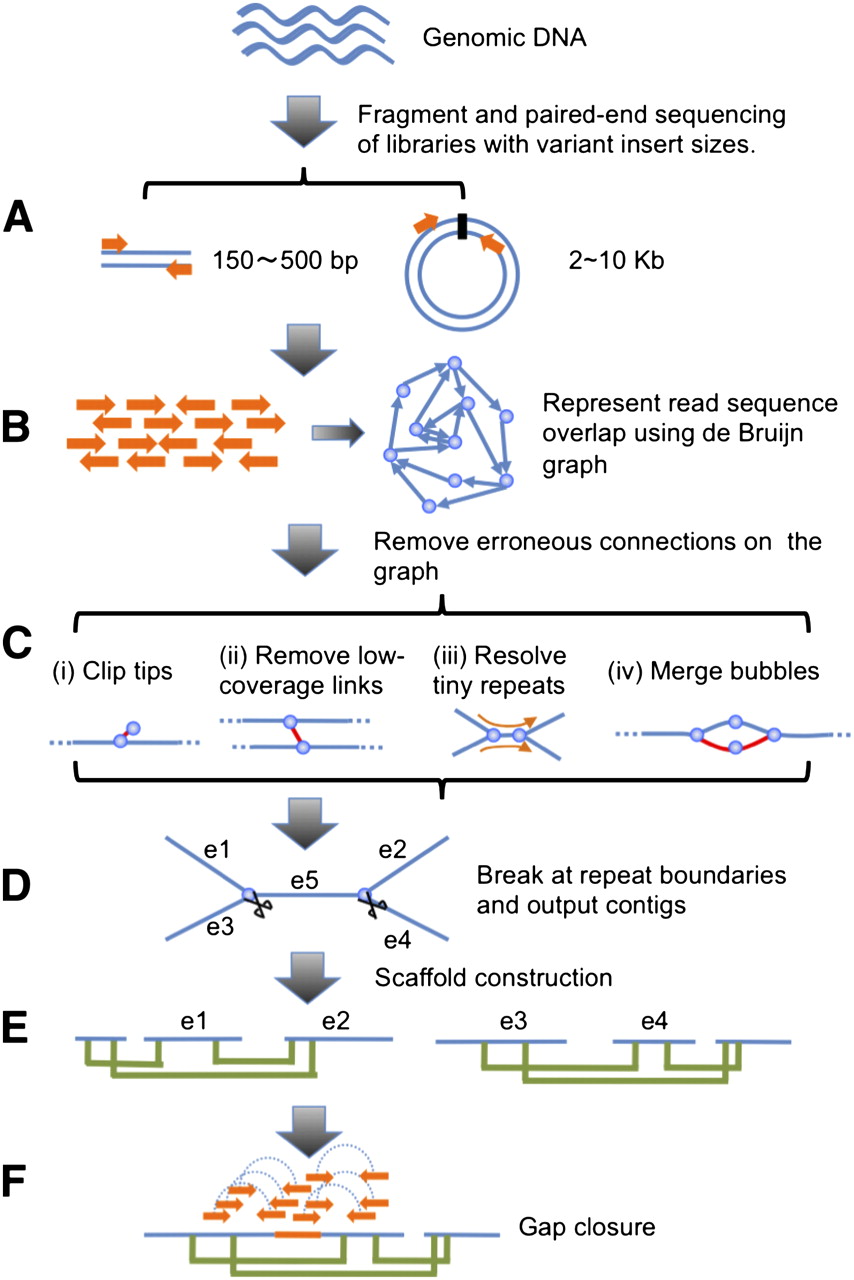
Once these erroneous edges were corrected, repeat connections on the graph were broken and linear sequences were output as contigs (Fig. 2D). By realigning the reads onto the contigs and transferring read pairing onto contig pairing relationships, we ordered the unique contigs and constructed them into scaffolds (Fig. 2E). Finally, the intrascaffold gaps were filled in through local assembly of the extracted reads inside the gap regions using paired-end information (Fig. 2F).
Detailed steps for genome assembly
Preassembly sequencing error correction
The rate of sequencing errors in Illumina GA reads is about 1%–2%. Even though errors primarily accumulate at the 3′-end of reads, many of the 25-mers will contain errors, which will make the total number of 25-mers much greater than expected. Error correction before assembly for small data sets is less important (and therefore optional) since the erroneous connections can easily be removed in the graph during assembly. This step, however, is essential for large data sets, as doing so tremendously reduces memory usage, making it feasible to load the complete number of read sequences and construct the de Bruijn graph.
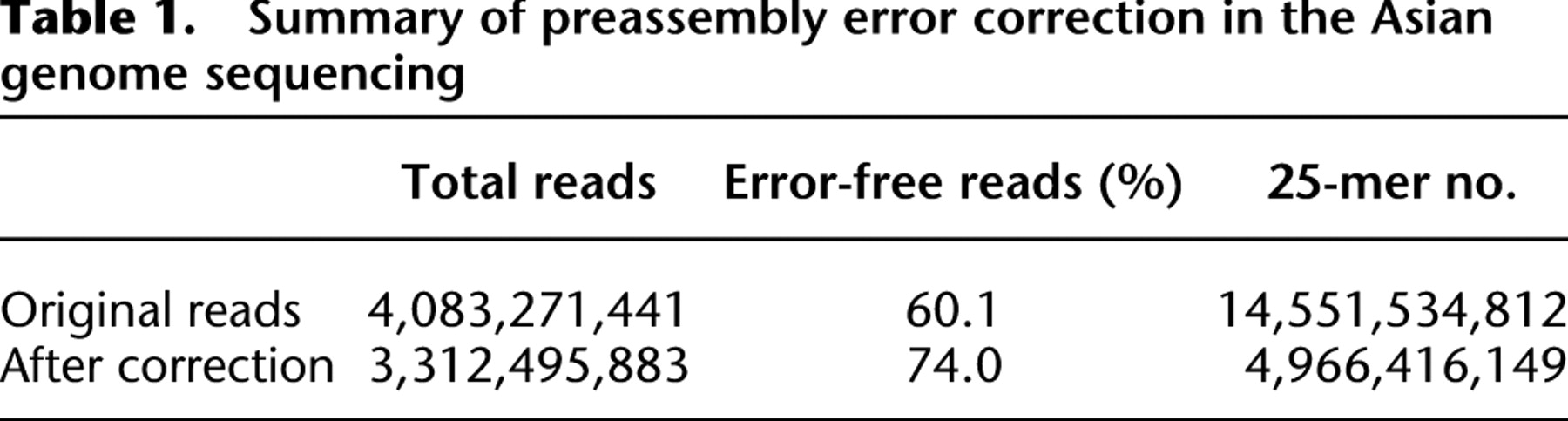
For the Asian genome data, the total number of distinct 25-mers was reduced from 14.6 billion to 5.0 billion (2.9 timers smaller) through this correction (Table 1). With the majority of the errors corrected, the ratio of error-free reads increased from 60.1% to 74.0%. A very small fraction (0.29%) of the reads might have been incorrectly revised in regions where sequence coverage was not deep enough, but these are unlikely to cause misassembly since paired-end information will be used in a later step to confirm the sequence overlap.
Summary of preassembly error correction in the Asian genome sequencing
Contig assembly
The initial de Bruijn graph was composed of 25-mers as nodes and the edge connection among the nodes was made up of read paths. We clipped the short tips that had lengths less than 50 bp in the graph. For the Asian genome, we removed 323.0 million (6.5%) tip nodes and also filtered 402.6 million low-coverage nodes that appeared only once, along with their related edges. Using read path information, we resolved 4.4 million tiny repeats. We merged 4.2 million bubbles that had a single base pair difference or two parallel paths that had less than a four base-pair difference, but had over 90% similarity into one path, and the higher-depth path was used to represent the common path. By reporting the contigs with lengths equal to or greater than 100 bp, the N50 and N90 sizes of the contigs were 1050 bp and 205 bp, respectively (Supplemental Table 2).
Scaffolding
After obtaining the contig sequences, we realigned the short reads onto the contigs. Since the repeat copies had been merged into consensus sequences in the graph and in the output contigs, each short read always mapped unambiguously to one contig. We used a minimum of three read pairs as the criteria to define the order and distance between two contigs, so the small fraction of chimeric reads will not create misassembly. Then the relationship among all the contigs was displayed as a graph. We masked the repeat contigs that had multiple and conflicting connections to the unique contigs. The remaining contigs with compatible connections to each other were linearized and constructed into scaffolds.
Starting from small and moving to larger insert sizes, we used the read mate pairs to join contigs into scaffolds step by step. By adopting 140- and 440-bp insert size paired-ends, the N50 of constructed scaffolds was 17.3 kb (Table 2). Adding 2.6 kb insert size paired-ends, the N50 size was improved to 103.5 kb. Adding 6- and 9.6-kb insert size libraries, the N50 of final scaffolds reached 446.3 kb. As shown in theoretical estimation, further improvement of N50 scaffold size depends on even larger insert size libraries.
Summary of the African and Asian genome assembly
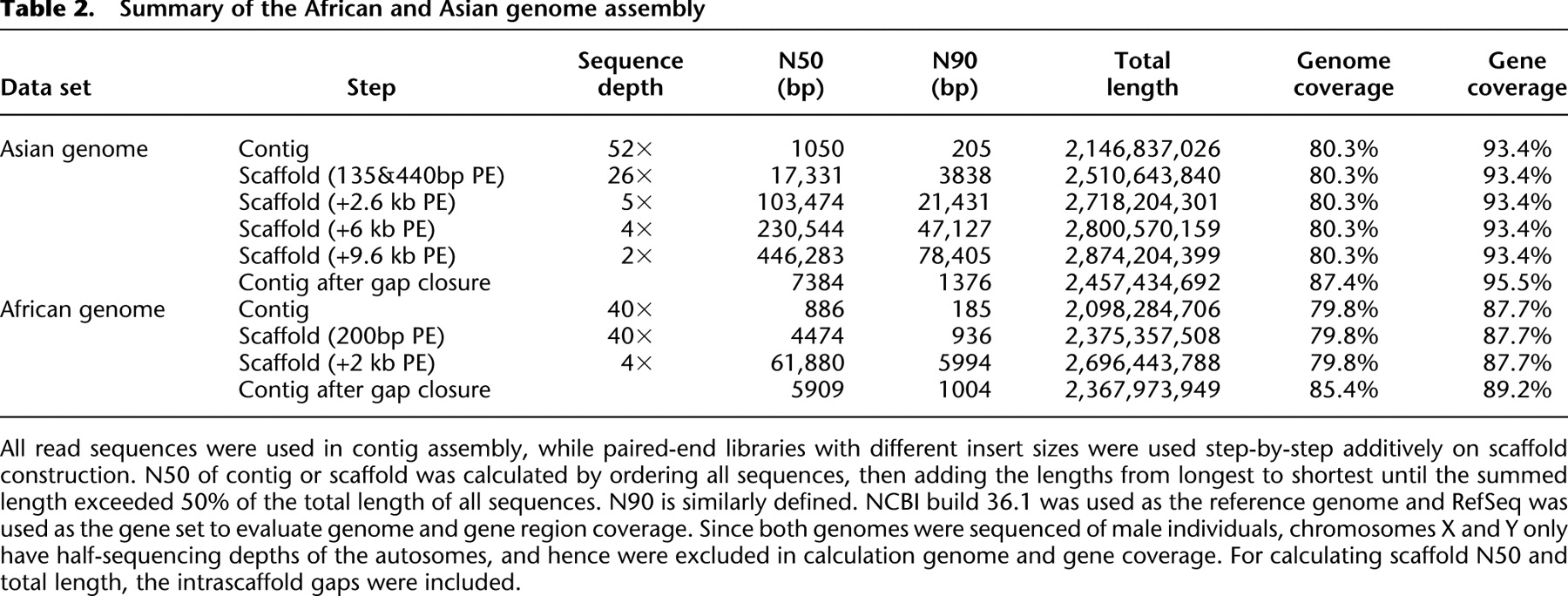
[i] All read sequences were used in contig assembly, while paired-end libraries with different insert sizes were used step-by-step additively on scaffold construction. N50 of contig or scaffold was calculated by ordering all sequences, then adding the lengths from longest to shortest until the summed length exceeded 50% of the total length of all sequences. N90 is similarly defined. NCBI build 36.1 was used as the reference genome and RefSeq was used as the gene set to evaluate genome and gene region coverage. Since both genomes were sequenced of male individuals, chromosomes X and Y only have half-sequencing depths of the autosomes, and hence were excluded in calculation genome and gene coverage. For calculating scaffold N50 and total length, the intrascaffold gaps were included.
Gap closure
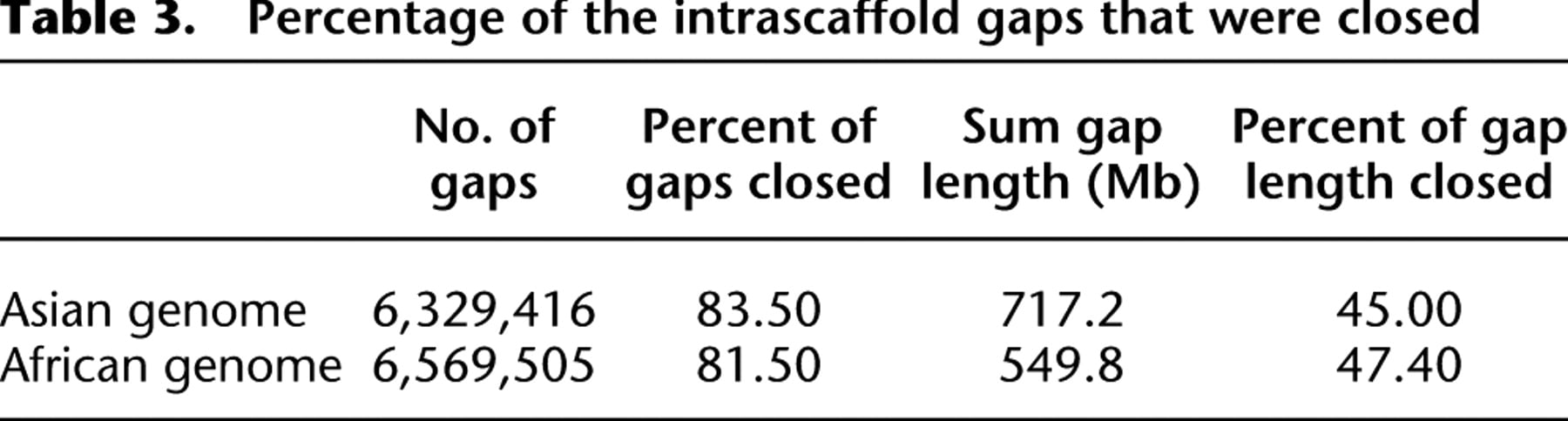
The majority of the gaps inside the scaffolds were composed of repeats that were masked during scaffold construction. To disassemble the repeat copies and fill in the gaps, we used the paired-end information to retrieve the read pairs that had one read well-aligned on the contigs and another read located in the gap region, then, a local assembly for the collected reads was done. We closed 83.5% of the 6.3 million intrascaffold gaps, or 45.0% of the 717-Mb sum gap length (Table 3). The contig N50 size grew from 1050 bp to 7.4 kb (if we ignore gaps <50 bp in length, the N50 size was 11.5 kb), and the genome coverage improved from 80.3% to 87.4% (Table 2).
Percentage of the intrascaffold gaps that were closed
Summary and comparison of the two assembled genomes
We applied the same assembly method to the African genome, and the final assembly of both genomes is summarized in Table 2. The assembly of the Asian genome had a longer N50 size than the African genome for contigs (7.4 kb vs. 5.9 kb) and for scaffolds (446.3 kb vs. 61.9 kb), which is likely due to the longer average read length (55 bp vs. 35 bp) and longer paired-end insert sizes (9.6 kb vs. 2 kb) of the Asian genome sequencing data.
Although there is structural variation between human genomes (Bentley et al. 2008; Kidd et al. 2008; Wang et al. 2008), comparison of the Asian and African genome assemblies against the NCBI human reference genome gave us a general assessment of genome coverage and assembly accuracy for both. The Asian sequence assembly had higher coverage of the NCBI reference genome than did the African genome (87.4% vs. 85.4%). This may be due to the Asian genome assembly having a longer total length, and may also be because the Asian genome is more similar to the NCBI reference genome than is the African genome (International HapMap Consortium 2007; Bentley et al. 2008; Wang et al. 2008). The lower coverage of the NCBI reference genome by the assembly method than by the mapping-based method (92% coverage) (Wang et al. 2008) can be explained by the fact that regions with insufficient sequence depth cannot be assembled and that small contigs (<100 bp) were filtered in the final assembly. According to the location of the RefSeq genes on the NCBI reference genome, the Asian and African genome assemblies covered 95.5% and 89.2% of the gene region, respectively.
Sequence accuracy of the assemblies
By mapping all the reads onto the assembled genomes, we calculated the allele frequency in the reads at each genomic location to measure assembly quality at a single-base level. The peak read depth of the Asian and African genome was 55 and 40, and over 99% of both assembled genomes had more than 20-read coverage (Supplemental Fig. 2). This indicated that deep short read sequencing has a higher base-level sequence accuracy than traditional Sanger sequencing, which normally has 4–10× coverage.
Since we previously annotated the SNPs between the Asian individual and the NCBI reference sequences (Wang et al. 2008), we aligned the Asian genome assembly with the NCBI reference to detect differing alleles and checked the overlap of these alleles to the previously identified SNPs in the Asian genome. There were 1.87 million mismatched alleles that comprised 0.09% of the aligned region. Only 78 alleles (0.004%) were inconsistent with the annotated SNPs.
Structural difference to the NCBI reference genome
From the comparison between the assembled genomes and the NCBI reference sequence, we observed structural differences that could be structural variations or misassemblies. To distinguish these two categories of differences and evaluate the rate of misassembly, we checked the number of supportive paired-ends and conflicting paired-ends to the assembly at the discrepant regions (Supplemental Fig. 3).
There were 2195 and 2406 contigs in the Asian and African genome that showed greater than 100-bp insertion or deletion against the NCBI reference sequence (Supplemental Table 3). The insert sizes of paired-ends were consistent with the span of their alignment on the assembly at these regions, so these insertions or deletions are more likely to be true. There were 117 and 3339 small contigs in the Asian and African assembly that failed to be placed into the gaps of scaffolds due to insufficient paired-end information at the regions. We found 3516 and 3339 contig insertions in Asian and African genomes compared to the NCBI reference. Only eight (0.2%) and three (0.1%) cases were potential misassemblies that have clearly more (over two times) conflicting than supportive paired-ends, while the others were true insertions. During the scaffolding process, some flanking pairs of small contigs may have been placed in an incorrect order because the contig sizes were in the same range of paired-end insert size deviation and were thus difficult to order. We observed 4715 and 3094 such cases in the Asian and African genomes, of which 1681 (35.7%) and 593 (19.2%) were likely misassembled. Including inversions and long-range translocations, the total length of discrepant regions were about 6 Mb and 5 Mb in the Asian and African genome, comprising 0.3% and 0.25% of the assembly, respectively.
Carrying out de novo assembly allowed us to identify small deletions and insertions; whereas, this is not possible by mapping-based methods when the length of deletion or insertion is comparable or smaller than the standard deviation of paired-end insert sizes. De novo assembly also has the advantage of resolving structural variations to a single-base level and obtaining the inserted sequences. Figure 3A shows an example of a detected 17-bp deletion. Figure 3B shows a detected 7926-bp insertion. Case-by-case analysis and further experiment validation will be required to fully characterize all of the structural variations.
Examples of deletion and insertion identified in the comparison of the assembled individual human genomes and the NCBI reference genome. (A) A 17-bp deletion in scaffold27122121 of the African genome located on chromosome 7. (B) A 7926-bp insertion in scaffold4928 of the Asian genome located on chromosome 7. The inserted sequence fragment was validated by a human BAC clone AC153461.2 in GenBank, and also exists in the chimpanzee genome.

Sequence depth effect on genome assembly
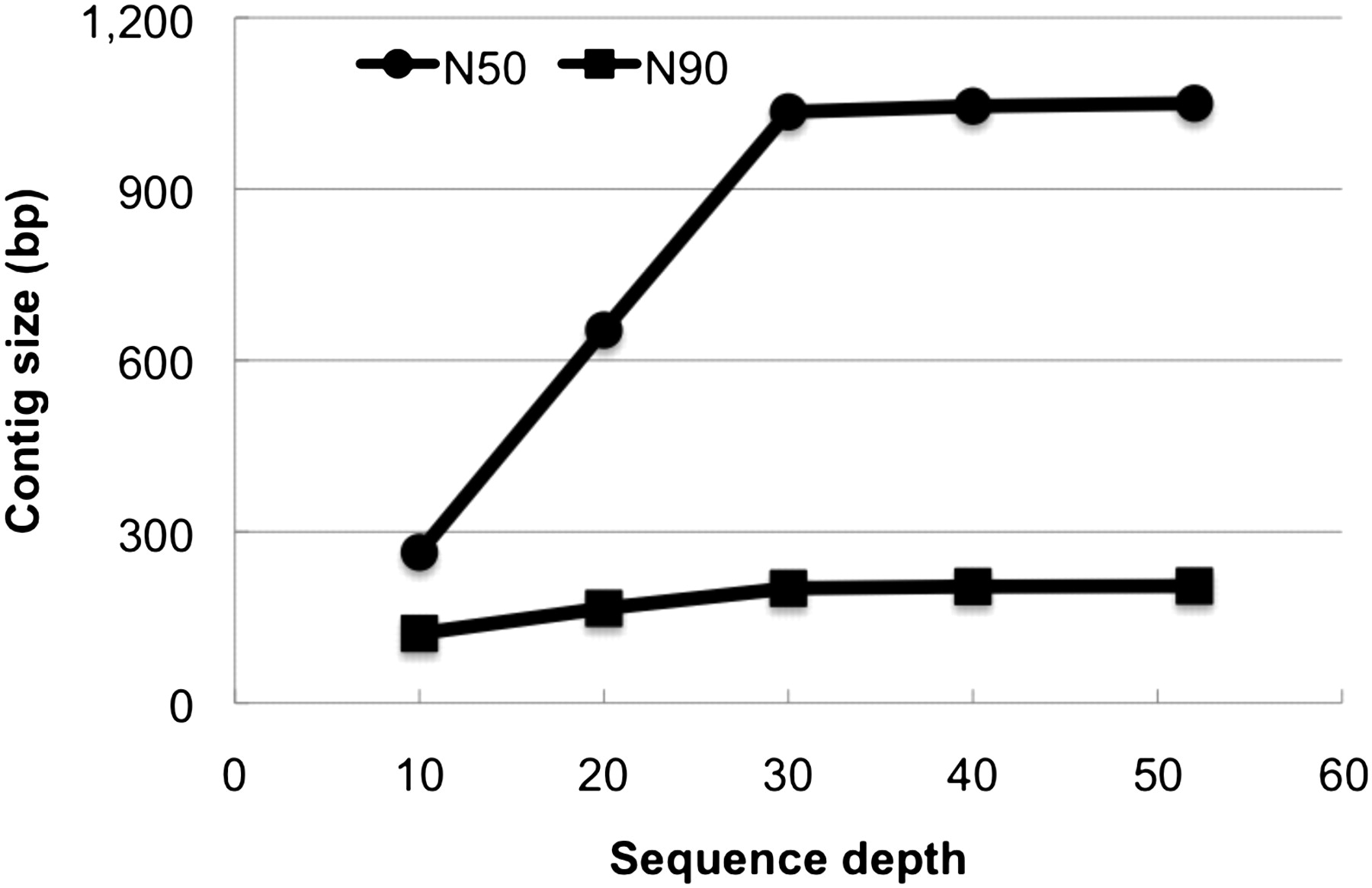
To determine the minimal sequence depth required for achieving a proper assembly of the human genome, we randomly sampled subsets of reads with different average depths from the error corrected reads of the Asian genome. This showed that contig size increased nearly linearly by improving sequence depth from 10× to 30×. Contig N50 and N90 size at 30× sequence depth were 1035 bp and 201 bp, respectively. We found that further improvement of sequence depth (to 40× and 50×) made only slight changes in contig size (N50 size of 1045 bp and 1050 bp) (Fig. 4). Accordingly, the total length of assembled contigs at 10×, 20×, and 30× sequence depth was 1.70, 2.09, and 2.15 Gb, respectively.
N50 and N90 size of assembled contigs by different sequence depths. We sampled subsets of randomly selected reads from the Asian genome data for de novo assembly of contigs. The same K-mer (K = 25) size was used for all the assemblies.
We also simulated reads with different lengths to investigate the optimal sequence depth for each read length. With a 35-bp read length, 30–50× provided the best results; with a 50-bp read length, 30× was best; and with a 75-bp read length, 20× sequence depth was sufficient (Supplemental Table 4). Considering that in real sequencing experiments DNA fragmentation is not completely random and there are unavoidable sequencing errors, we suggest that 5–10× more reads than the theoretical estimate would be best for achieving optimal assembly.
As was shown in Figure 1B and Table 1, the length of paired-end insert sizes determined the scaffold size of de novo genome assembly. SOAPdenovo required a minimum of 50× physical clone coverage for each of the libraries of ∼200 bp, ∼500 bp, ∼2 kb, ∼5 kb, ∼10 kb, etc.
Computational complexity and comparison to the other assemblers
We determined the memory usage and computational complexity of SOAPdenovo by the size and repeat abundance of the sequenced genome and by the sequencing quality. We assembled the two human genomes on a supercomputer of eight Quad-core AMD 2.3 GHz CPUs with 512 Gb memory installed. For the computational intensive steps, we used threaded parallelization. The preassembly error correction of the raw reads was the most time consuming step, which cost 24 and 22 h, respectively, on the Asian and African data set. The de Bruijn graph construction step had the highest peak memory usage (140 Gb). In total, we finished assembly of the Asian and African genomes within 48 h and 40 h, respectively, on the supercomputer (Table 4).
Statistics of computational complexity at each assembly step
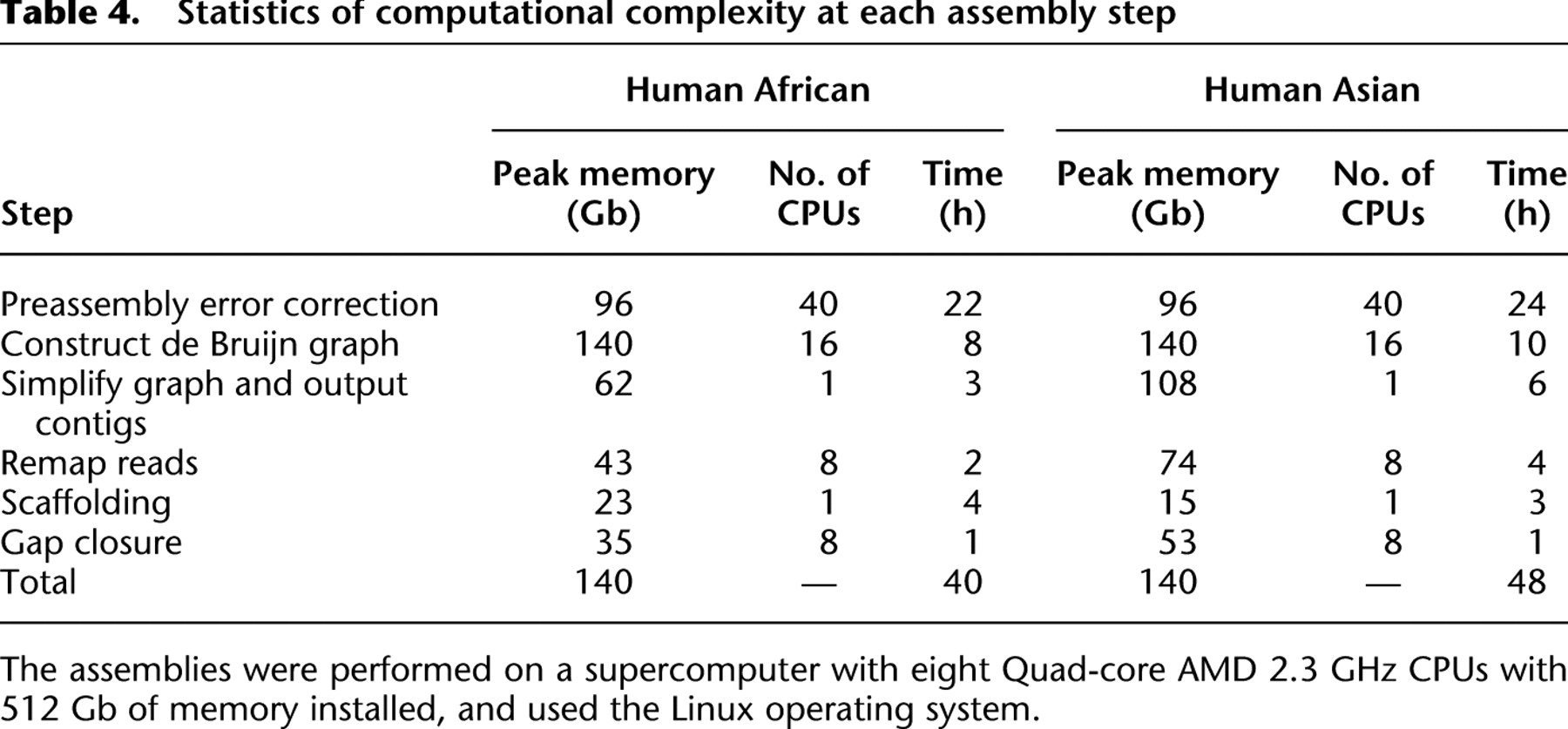
[i] The assemblies were performed on a supercomputer with eight Quad-core AMD 2.3 GHz CPUs with 512 Gb of memory installed, and used the Linux operating system.
ABySS (Simpson et al. 2009) was tested on the same African genome data set that was used here, with the only difference being that ABySS used 42× data of 210-bp insert-size libraries, while we included an additional 4× paired-end reads from a ∼2-kb insert-size library in this analysis. To provide a fair comparison, we ran SOAPdenovo excluding the additional 4× reads, and obtained a contig N50 length of 4611 bp and 85% coverage of the NCBI human reference genome, which is significantly better than ABySS (contig N50 size of 1499 bp and genome coverage of 68%). SOAPdenovo finished the assembly within 40 h on a 32-core (2.3 GHz CPU) supercomputer, while ABySS used 87 h on a 168-core (2.66 GHz CPU) Linux cluster. This indicated that SOAPdenovo is also faster than ABySS. However, as has been noted, SOAPdenovo has a much higher peak memory usage (140 Gb) than ABySS (<16 Gb).
To evaluate and compare the performance of SOAPdenovo to other available short read assemblers, we tested the program on the data set of 20.8 M paired-end 36-bp Illumina GA reads generated from a 200-bp insert size of E. coli library (SRX000429); the other assemblers, including ABySS (Simpson et al. 2009), Velvet (Zerbino and Birney 2008), EULER-SR (Chaisson and Pevzner 2008), SSAKE (Warren et al. 2007), and Edena (Hernandez et al. 2008), have been evaluated using this data set (Simpson et al. 2009). SOAPdenovo reported 182 contigs (>100 bp in length), with mean size of 25 kb and N50 size of 89 kb, and only five incorrect contigs, which indicated both longer contig size and assembly accuracy than the others. Further, SOAPdenovo connected the contigs into 148 scaffolds with N50 size of 105 kb.
Discussion
The short read de novo assembly methods we have developed make it possible for building reference genome sequences for novel species in a more efficient and cost-effective way. Currently, using the Illumina GA sequencing technology and our short read assembler presented here, we have sequenced and assembled nearly a dozen plant and animal genomes, including the panda (Li et al. 2009d), duck, potato, cucumber (Huang et al. 2009), watermelon, and others.
Repeat characteristics in different genomes can vary extensively, so the expected theoretical de novo assembly results from different genomes will also vary. A similar theoretical assessment in Drosophila, mouse, and rice genomes showed that the expected contig N50 size of unique contigs by using 25-mer as seed is 5.9, 1.1, and 1.2 kb, respectively (Supplemental Figs. 4–6). The Drosophila euchromatic genome region contains relatively fewer repeats, thus, with a 200-bp paired-end insert size, we would expect to achieve a 110-kb scaffold N50 size. While for the rice genome, which contains numerous long retrotransposons, we would only expect a 4.2-kb scaffold N50 size by using 200-bp paired-ends and 96 kb by including up to 5-kb paired-end insert sizes (Supplemental Figs. 7–9). A survey of the repeat characteristics of phylogenetically closer genomes will provide guidance in designing experiments, and setting goals for optimizing the assembler for novel genomes. But in practice, it is very difficult to reach the theoretical limit for every potential problem that may occur during the sequencing process, including biased DNA fragmentation, sequencing errors, inaccurate paired-end insert sizes, and others.
Longer reads will help improve contig size, while long insert-size libraries will be essential for crossing repeat clusters and construction of long scaffolds. In theory, without long insert-size libraries, repeats that extend beyond the paired-end insert sizes will not be able to be resolved and assembled. For gap closure, the last step in assembly, sufficient sequencing depth of each insert-sized library is correlative to the effectiveness of filling the corresponding sized gaps.
In our method, we used a similar de Bruijn graph data structure as Velvet (Zerbino and Birney 2008), but we did not record the read locations and paired-end information in the graph as is done in Velvet. This made it feasible to build a graph using a complete, and very large, read set of the whole human genome. The modularized pipeline format of SOAPdenovo also has the advantage of easy modification or addition for further development and improvement.
The ability to decode the genomes of all major evolutionary clades and any additional useful or interesting organisms would tremendously broaden our knowledge of evolutionary mechanisms, help determine complete gene sets, detect biological underpinnings of diseases, and more. In addition to haploid or homogeneous diploid genome assembly, short read sequencing technologies can, in principle, also be used for highly heterogeneous diploid or polyploid genome assembly, metagenomics data assembly, and large-scale transcriptome data assembly. But this will require novel data format definition to properly present the assembly results, and thus remains one of the biggest challenges for developing practical methods.
Methods
Genome data
The reference sequence used was NCBI build 36.1, and the gene set of the human genome used was from RefSeq. Both the chromosome sequences and the gene set were downloaded from the UCSC database (http://genome.ucsc.edu/). The sequenced African individual (Bentley et al. 2008) was a male Yoruba (NA18507), from the HapMap samples. The Asian genome data was from a male Han Chinese (Wang et al. 2008). Both samples were sequenced by Illumina Genome Analyzer (GA) technology, and the data sets are freely available at the EBI/NCBI Short Read Archive with the following accession numbers: African, SRA000271; Asian, ERA000005.
Error correction
For deep sequencing, the correct K-mers appear multiple times in the reads set, while random sequencing error-containing K-mers have low frequency. Our error correction method used K-mer frequency information. The K-mer size was determined by the genome size, read length, and supercomputer memory. Since we only need to correct the low-frequency K-mer, to save memory, we used one byte for each K-mer to store the frequency and assign all counts over 255 as 255. Here, we chose K = 17 bp, because 417 = 16 G is larger than the genome size; thus, error-containing 17-mers were unlikely to exist in real genomes. The peak frequency of correct 17-mers would be about 20 in the read sets. We built a hash table to store the frequency of all 17-mers, which occupied 16 Gb of memory. Then, for each read, we started from high-frequency regions and extended both sides to infer potential erroneous sites of low-frequency (<3) 17-mers. For each inferred erroneous site, we tested the impact of changing it to the other three allele types, and these changes were picked up as candidates if all 17-mers containing the allele had a frequency equal to or over 3. If we obtained no candidates that satisfied these criteria, we did not change it; otherwise, the allele was revised to that with the highest 17-mer frequency. A dynamic programming algorithm was used to find the optimal solution with minimal changes. To increase speed, we used threaded parallelization to split read sets and handled them in parallel by sharing the same 17-mer hash table.
De Bruijn graph construction
For the de Bruijn graph, each node is a K-mer. Two nodes that overlap K − 1 bp and appeared in a neighboring read sequence were connected as an edge. Small K-mers make the graph very complex with a lot of edges created by repeat sequences; while large K-mers can have poor overlap in regions with low sequencing depth. After assessing different K-mer sizes, we found that the 25-mer provided the best tradeoff.
Tip removal
A single base-pair sequencing error on a read will create K consecutive incorrect K-mers. If the error occurs in the middle of a read and the K-mers at both ends are correct, the path created by the error would appear as a bubble (the bubble is discussed below) in the graph; otherwise, it would cause a “dead end,” or a tip, in the graph. We removed the tips that were shorter than 2K (50 bp if K = 25) and had a lower frequency than other alternative paths that connected at a common destination node in the graph.
Solving tiny repeats
Tiny repetitive sequences in the graph that are shorter than the read length may be able to be resolved by read paths. To avoid misassembly, we only tried to solve repeat nodes with equal N incoming and outgoing edges. If each of the N incoming edges had read path support from one of the N outgoing edges and had no conflicts, we then removed the repeat node and split the connections into N parallel paths.
Merging bubbles
We used Dijkstra's algorithm to detect bubbles, which is similar to the “Tour-bus” method in Velvet. We merged the detected bubbles into a single path if the sequences of the parallel paths were very similar; that is, only had a single base pair difference or had fewer than four base pairs difference with >90% identity.
Contig linkage graph
The first step of scaffolding is to realign the reads onto the contig sequences. Then the paired-end relationship between the reads was transferred to linkage between contigs. The linkages among all contigs formed a graph. We used the number of read pairs between two contigs to weight the linkage, and used the read paired-end insert sizes to estimate the gap size between the two contigs. Theoretically, if the insert size of a paired-end clone library obeys Normal distribution with a mean value μ and variance σ2, the gap size estimated from N paired-ends will also have mean μ, but variance σ2/N. For our method, we required as least three read pairs to form a linkage.
Scaffolding
We used two steps to simplify the contig linkage graph and extract unambiguously linear paths from the graph to construct scaffolds. The first step is subgraph linearization: The compatible transitive lineages among a group of contigs were removed and the contigs were merged into one node with carefully estimated internal gap sizes. The next step is repeat masking: If a contig has multiple incoming and outgoing linkages to the other contigs, but the linkages are not compatible, we defined the contig as a repeat. The repeat contigs, together with their linkages, were masked during scaffolding. Since we have multiple paired-end read sets with different insert sizes, and clone physical coverage for each insert-sized read set is very deep and sufficient for reliable scaffold construction, we constructed scaffolds starting with short paired-ends and then iterated the scaffolding process, step by step, using longer insert size paired-ends. This strategy effectively made scaffold construction easier while avoiding interleaving.
Acknowledgments
This project is supported by the Chinese Academy of Science (GJHZ0701-6; KSCX2-YWN-023), the National Natural Science Foundation of China (30725008; 30890032; 90608010, 30811130531), the Chinese 973 program (2007CB815701; 2007CB815703; 2007CB815705), the Chinese 863 program (2006AA02Z177; 2006AA10A121), the International Science and Technology Cooperation Project (0806), the Shenzhen hundred-million project, the Danish Platform for Integrative Biology, the Ole Rømer grant from the Danish Natural Science Research Council, the pig bioinformatics grant from Danish Research Council, the Solexa project (272-07-0196), the Danish Strategic Research Council grant no. 2106-07-0021 (Seqnet), and the Lundbeck Foundation Centre of Applied Medical Genomics for Personalized Disease Prediction, Prevention and Care (LUCAMP). We thank Laurie Goodman for editing the manuscript.
References
- ↵S BatzoglouDB JaffeK StanleyJ ButlerS GnerreE MauceliB BergerJP MesirovES Lander(2002) ARACHNE: A whole-genome shotgun assembler. Genome Res 12:177–189.
- ↵DR Bentley(2006) Whole-genome re-sequencing. Curr Opin Genet Dev 16:545–552.
- ↵DR BentleyS BalasubramanianHP SwerdlowGP SmithJ MiltonCG BrownKP HallDJ EversCL BarnesHR Bignell(2008) Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456:53–59.
- ↵J ButlerI MacCallumM KleberIA ShlyakhterMK BelmonteES LanderC NusbaumDB Jaffe(2008) ALLPATHS: De novo assembly of whole-genome shotgun microreads. Genome Res 18:810–820.
- ↵MJ ChaissonPA Pevzner(2008) Short read fragment assembly of bacterial genomes. Genome Res 18:324–330.
- ↵JC DohmC LottazT BorodinaH Himmelbauer(2007) SHARCGS, a fast and highly accurate short-read assembly algorithm for de novo genomic sequencing. Genome Res 17:1697–1706.
- ↵TD HarrisPR BuzbyH BabcockE BeerJ BowersI BraslavskyM CauseyJ ColonellJ DimeoJW Efcavitch(2008) Single-molecule DNA sequencing of a viral genome. Science 320:106–109.
- ↵P HavlakR ChenKJ DurbinA EganY RenXZ SongGM WeinstockRA Gibbs(2004) The Atlas genome assembly system. Genome Res 14:721–732.
- ↵D HernandezP FrancoisL FarinelliM OsterasJ Schrenzel(2008) De novo bacterial genome sequencing: Millions of very short reads assembled on a desktop computer. Genome Res 18:802–809.
- ↵X HuangJ WangS AluruSP YangL Hillier(2003) PCAP: A whole-genome assembly program. Genome Res 13:2164–2170.
- ↵S HuangR LiZ ZhangL LiX GuW FanWJ LucasX WangB XieP Ni(2009) The genome of the cucumber, Cucumis sativus L. Nat Genet 41:1275–1281.
- ↵International HapMap Consortium (2007) A second generation human haplotype map of over 3.1 million SNPs. Nature 449:851–861.
- ↵WR JeckJA ReinhardtDA BaltrusMT HickenbothamV MagriniER MardisJL DanglCD Jones(2007) Extending assembly of short DNA sequences to handle error. Bioinformatics 23:2942–2944.
- ↵JM KiddGM CooperWF DonahueHS HaydenN SampasT GravesN HansenB TeagueC AlkanF Antonacci(2008) Mapping and sequencing of structural variation from eight human genomes. Nature 453:56–64.
- ↵ES LanderLM LintonB BirrenC NusbaumMC ZodyJ BaldwinK DevonK DewarM DoyleW FitzHugh(2001) Initial sequencing and analysis of the human genome. Nature 409:860–921.
- ↵R LiY LiK KristiansenJ Wang(2008) SOAP: Short oligonucleotide alignment program. Bioinformatics 24:713–714.
- ↵R LiY LiH ZhengR LuoH ZhuQ LiW QianY RenG TianJ Li(2009a) Building the sequence map of the human pan-genome. Nat Biotechnol 10.1038/nbt.1596.
- ↵R LiY LiX FangH YangJ WangK KristiansenJ Wang(2009b) SNP detection for massively parallel whole-genome resequencing. Genome Res 19:1124–1132.
- ↵R LiC YuY LiTW LamSM YiuK KristiansenJ Wang(2009c) SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 25:1966–1967.
- ↵R LiW FanG TianH ZhuL HeJ CaiQ HuangQ CaiB LiY Bai(2009d) The sequence and de novo assembly of the giant panda genome. Nature 10.1038/nature08696.
- ↵Mouse Genome Sequencing Consortium (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420:520–562.
- ↵JC MullikinZ Ning(2003) The phusion assembler. Genome Res 13:81–90.
- ↵EW MyersGG SuttonAL DelcherIM DewDP FasuloMJ FlaniganSA KravitzCM MobarryKH ReinertKA Remington(2000) A whole-genome assembly of Drosophila. Science 287:2196–2204.
- ↵PA PevznerH TangMS Waterman(2001) An Eulerian path approach to DNA fragment assembly. Proc Natl Acad Sci 98:9748–9753.
- ↵J ShendureRD MitraC VarmaGM Church(2004) Advanced sequencing technologies: Methods and goals. Nat Rev Genet 5:335–344.
- ↵JT SimpsonK WongSD JackmanJE ScheinSJ JonesI Birol(2009) ABySS: A parallel assembler for short read sequence data. Genome Res 19:1117–1123.
- ↵JC VenterMD AdamsEW MyersPW LiRJ MuralGG SuttonHO SmithM YandellCA EvansRA Holt(2001) The sequence of the human genome. Science 291:1304–1351.
- ↵J WangGK WongP NiY HanX HuangJ ZhangC YeY ZhangJ HuK Zhang(2002) RePS: A sequence assembler that masks exact repeats identified from the shotgun data. Genome Res 12:824–831.
- ↵J WangW WangR LiY LiG TianL GoodmanW FanJ ZhangJ LiJ Zhang(2008) The diploid genome sequence of an Asian individual. Nature 456:60–65.
- ↵RL WarrenGG SuttonSJ JonesRA Holt(2007) Assembling millions of short DNA sequences using SSAKE. Bioinformatics 23:500–501.
- ↵DR ZerbinoE Birney(2008) Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829.
Notes
[3] [Supplemental material is available online at http://www.genome.org. SOAPdenovo is freely available at http://soap.genomics.org.cn/soapdenovo.html. The genome assembly results for the Asian and African individuals have been submitted to GenBank (http://www.ncbi.nlm.nih.gov/Genbank/) under accession nos. ADDF000000000 and DAAB000000000, respectively. The versions described in this study are the first versions, ADDF010000000 and DAAB010000000. The assembly and analysis results are also available at http://yh.genomics.org.cn.]
[4] Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.097261.109.