
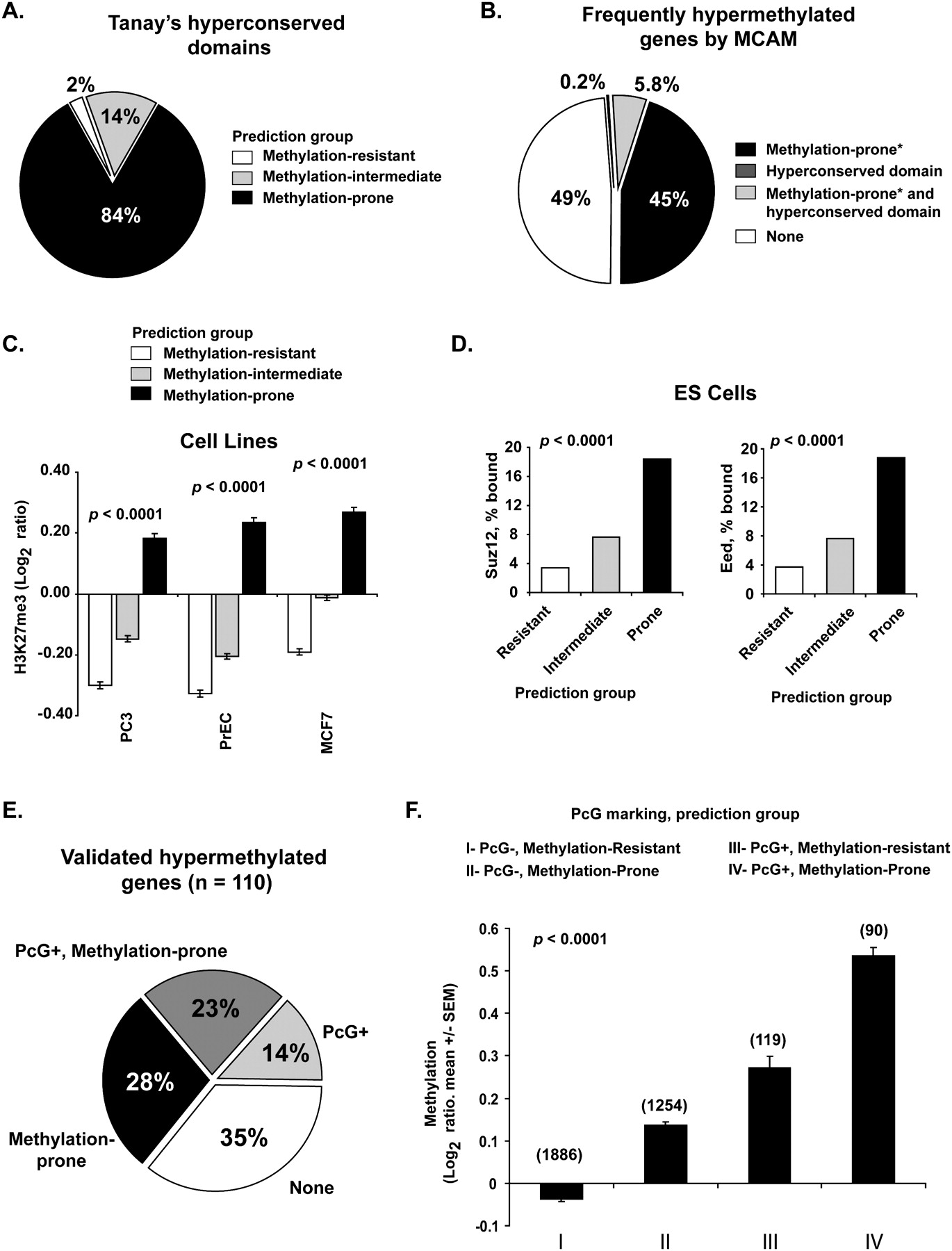
Genome architecture influences on PcG protein binding in embryonic and differentiated cells. (A) Frequency of predicted methylation groups among hyperconserved domains. (B) Relative contribution of hyperconserved domains and retrotransposon depletion in marking frequently methylated genes in cancer. MCAM data from 32 primary tissues and 28 cancer cell lines were averaged to identify frequently methylated genes. *Predicted status. (C) Enrichment of H3K27me3 mark in predicted methylation-prone genes in cancer (PC3, prostate; MCF7, breast) and normal immortalized (PrEC, prostate epithelium) cell lines. H3K27me3 marking was measured by ChIP with microarray hybridization (ChIP-chip) and is quantified as log2 ratio of pull-down signal over no antibody signal (Kondo et al. 2008). (D) Frequency of binding of SUZ12 and EED (PcG proteins) in human embryonic stem cells to 2583 methylation-resistant, 3655 methylation-intermediate, and 1690 methylation-prone genes based on our predictive model. Note that genes predicted methylation-prone (thus depleted for SINE and LINE retrotransposons) are preferential targets of PcG proteins. (E) Comparison of PcG marking and our predictive model in identifying methylation-prone genes from our training and first testing set. (F) Average measured methylation of predicted methylation-prone and methylation-resistant genes in PcG marked genes. MCAM data from 32 primary tumors and 28 cancer cell lines were averaged per comparison group, and methylation is presented as log2 ratio (cancer/control). The number of genes per category is presented above each column.








