
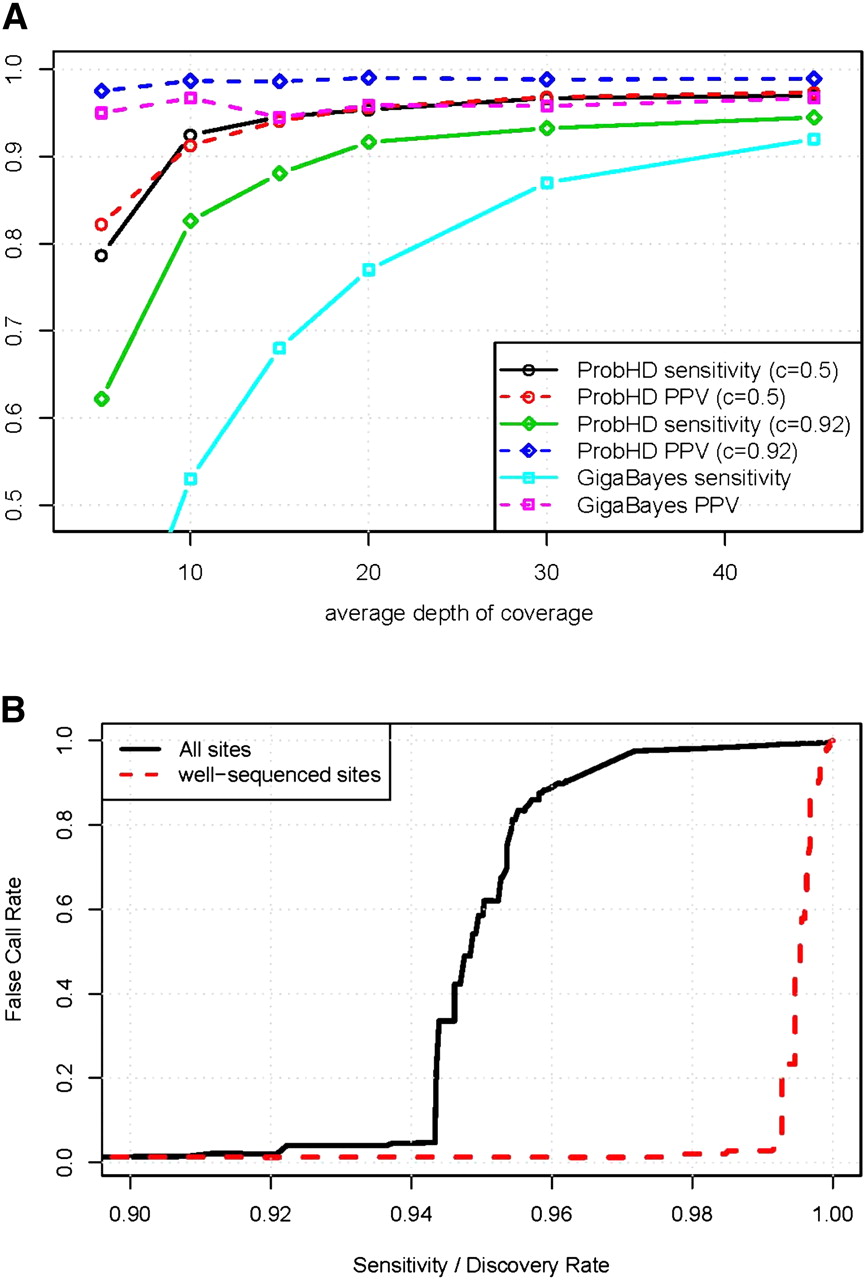
(A) Effect of coverage depth on prediction of known heterozygous sites. Sensitivity and positive predictive value (PPV, equal to 1 − FCR) are shown as a function of average depth of coverage. ProbHD results are shown with two different probability cutoffs for predicting heterozygous sites. A cutoff of c = 0.92 yields a conservative predictor that makes few false-positives, and a cutoff of c = 0.5 yields a very liberal predictor with higher sensitivity but higher FCR. Results are not corrected for HapMap errors. (B) Estimated de novo SNP-calling sensitivity and FCR, assuming 0.1% of sites are heterozygous. Well-sequenced sites are those sites with at least 13× coverage that are located on amplicons with minor allele deriving at least 25% of reads. The pronounced “elbow” is due to the severe imbalance between heterozygous and homozygous sites. Using a very conservative confidence threshold yields an error rate close to zero. However, as the threshold is lowered the percentage of homozygous sites miscalled as heterozygous sites eventually becomes nonzero. Even when the percentage of errors is quite small, the absolute number of errors quickly becomes large in comparison to the number of true hets, and the FCR climbs rapidly.








