
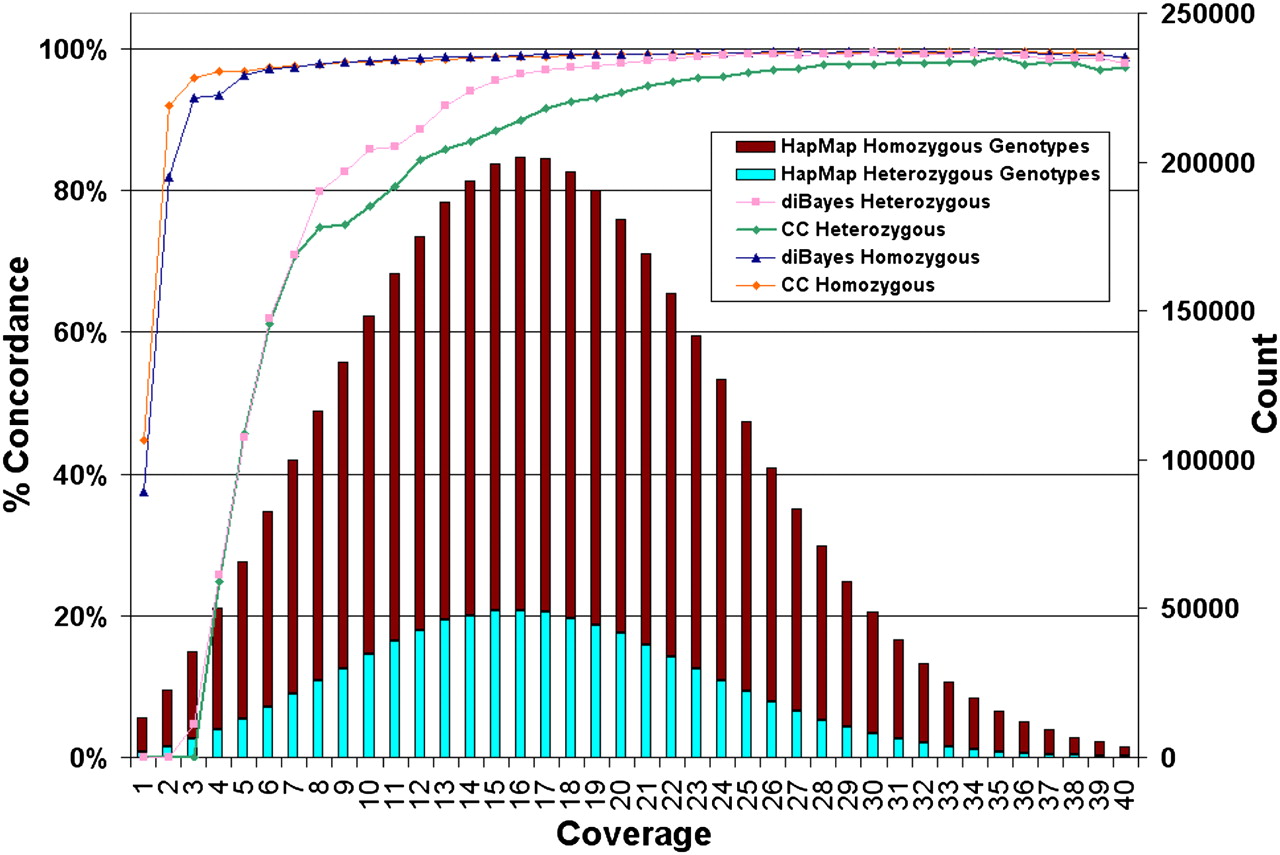
Dependence of genotype calling on depth of sequence coverage. The NA18507 genotypes called by SOLiD at all HapMap loci are compared with the HapMap genotypes by SOLiD coverage per genome position (average 18× coverage). Coverage includes alleles representing the reference or a valid base change; i.e., alleles with single or invalid adjacent mismatches are not included. No prior information about SNP presence or SNP alleles was used in making SOLiD gentoype calls. The number of HapMap loci with a given level of SOLiD coverage (“Count”) are shown and the percentage of these loci for which SOLiD gives the same genotype as HapMap for homozygotes and heterozygotes is represented by the colored lines (graphed using the left-hand y-axis and referred to as “% Concordance”) using two genotyping algorithms: Consensus Caller and diBayes. diBayes is more sensitive at heterozygous SNP detection and yields a lower false-negative rate than Consensus Caller, but we did not attempt to estimate the false-positive rate of diBayes with validation data. SOLiD genotypes that differ from HapMap gentoypes are nearly always heterozygous undercalls (i.e., the position is called homozogyous for one of the two alleles) or called as N (insufficient evidence to make a confident genotype call).








