
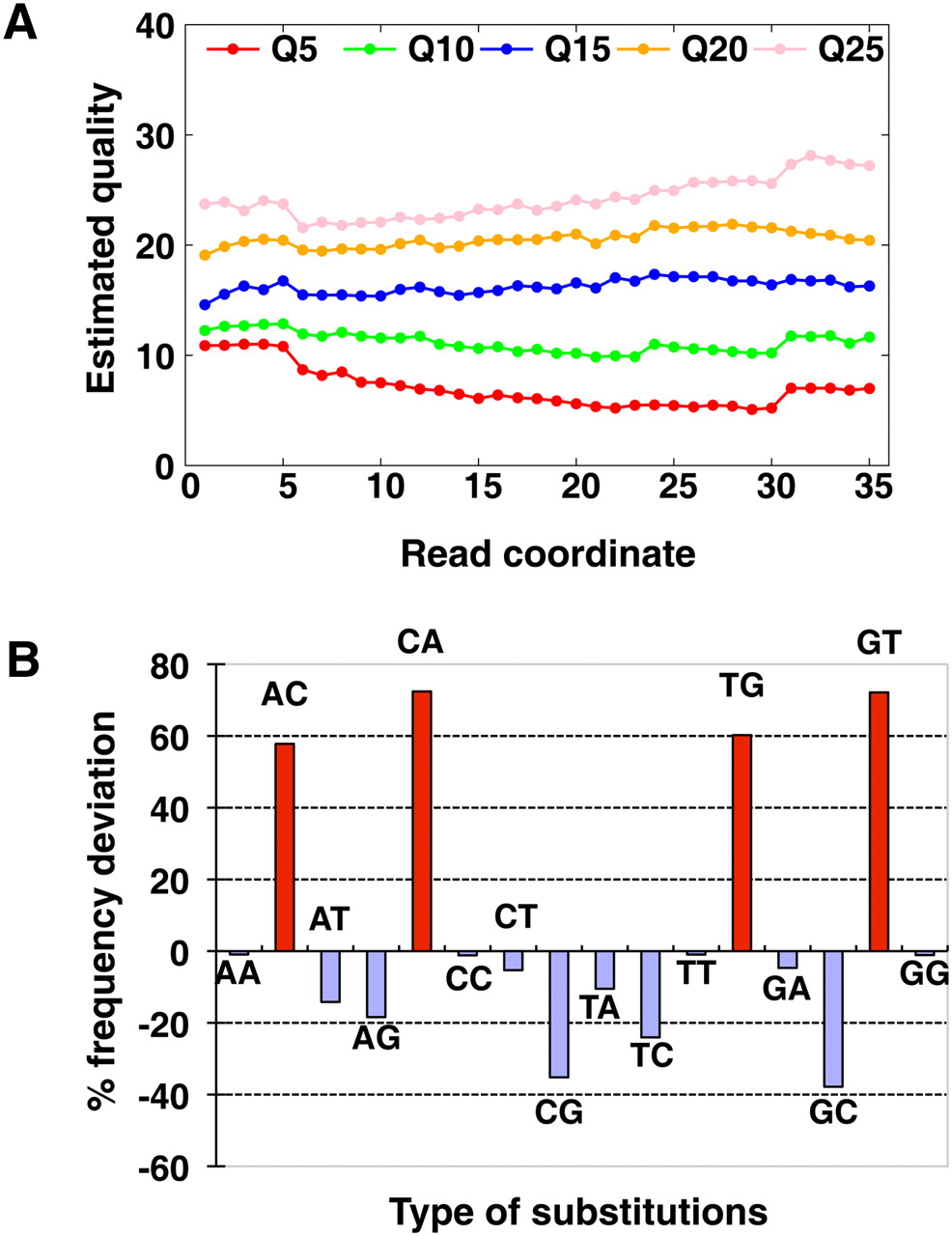
Inaccuracy of sequencing quality score and biased substitution errors. (A) Estimated quality of Illumina GA pipeline recalibrated quality values 5, 10, 15, 20, and 25 along sequencing cycles. We extracted bases with each quality value from raw reads of the Asian genome sequencing, then estimated the real quality by the mismatch rate in the alignment as [−10log10(mismatch rate)]. (B) Deviation of quality score to estimated mismatch rate of each substitution combination. The percentage of deviation was calculated by [(Error rate by alignment mismatch rate) − (Error rate according to quality value)]/(Error rate according to quality value). The substitution of A on read to C on reference was represented as AC in the figure.








