
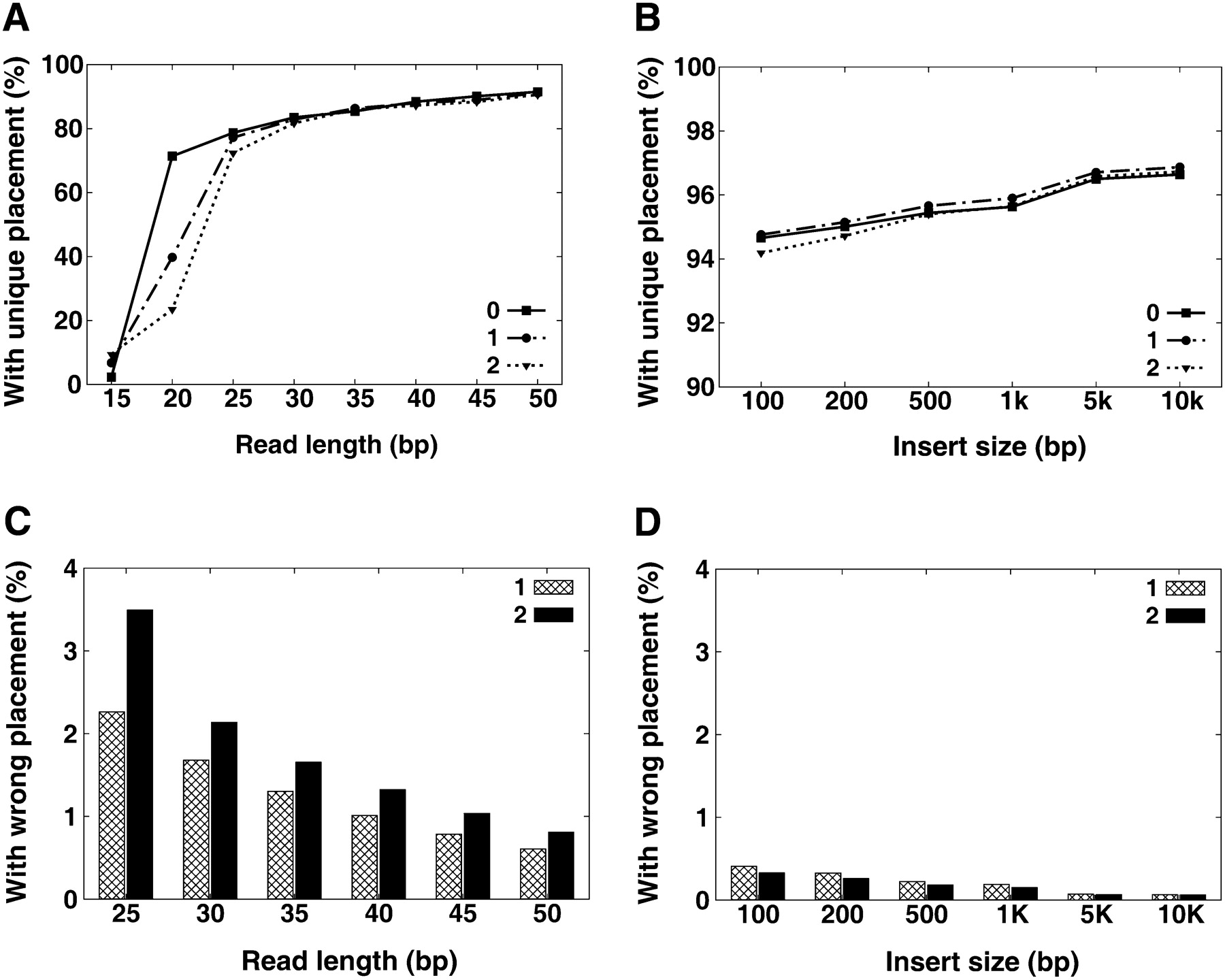
Uniqueness and accuracy of read placement. We produced a copy of human chromosome 12 with a 0.001 SNP rate to the NCBI reference, then simulated 36× reads for each given read length or paired-end insert size. The simulated error rate over all reads is ∼1%, and the standard deviation on paired-end insert size is 10%. (See Methods section for details.) All the read sequences were then aligned back to the reference genome, and then the uniqueness and accuracy of reads placement was evaluated: (A) unique placement of single-end reads; (B) unique placement of paired-end reads (read length: 35 bp); (C) wrong placement of single-end reads; (D) wrong placement of paired-end reads (read length, 35 bp).








