
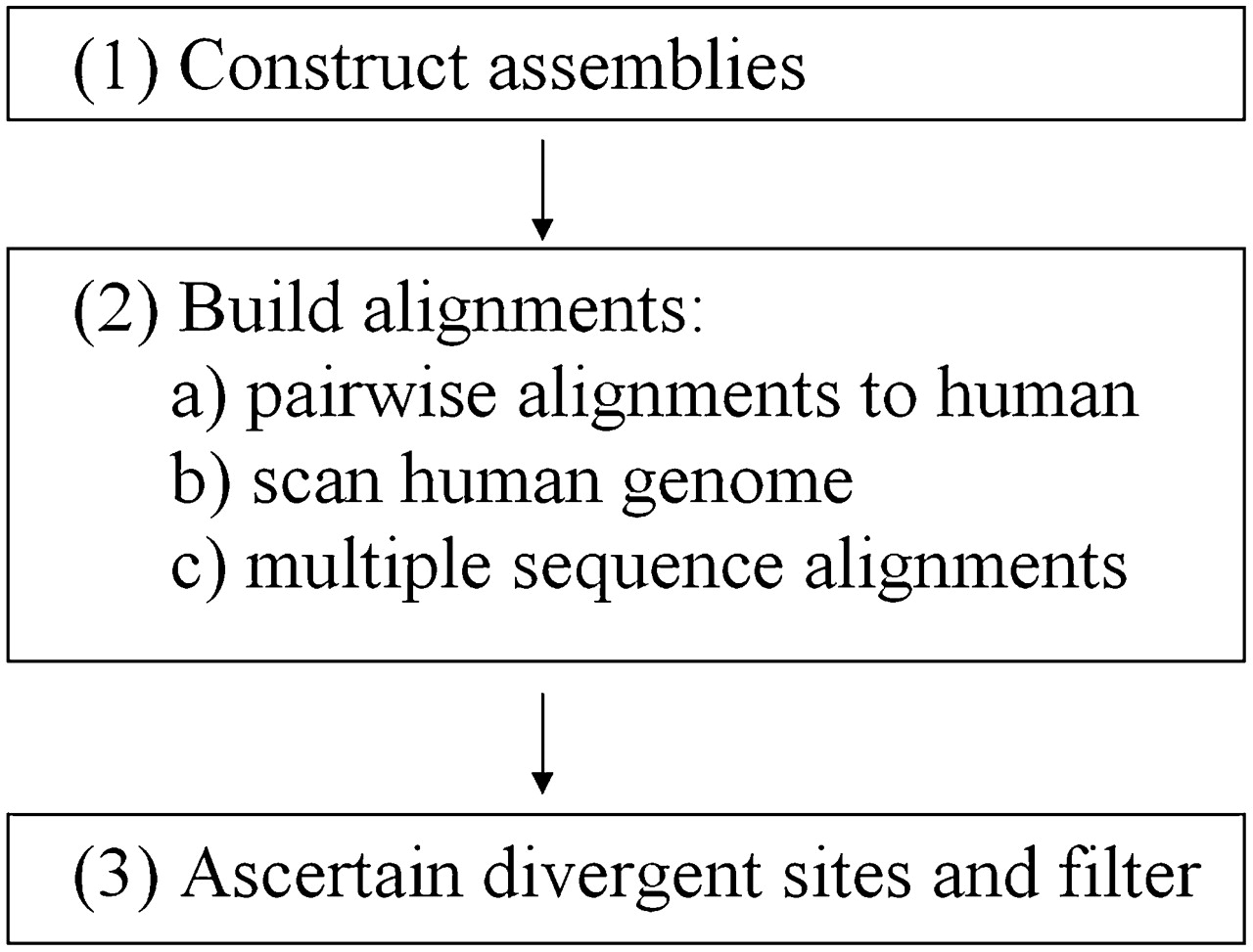
Alignment pipeline. Flowchart of our bioinformatic procedure for generating multiple sequence alignments. For non-human species in step 1, publicly available traces are turned into genome assemblies using ARACHNE (Jaffe et al. 2003). This allows us to construct a synteny map and to use assembly information to guide the positioning of the non-human sequence on the reference (human) genome. In step 2, pairwise alignments of non-human sequence with its human counterpart are constructed using synteny information and information on the uniqueness of the alignment to filter out spurious alignments and regions of duplication. BLASTZ (Schwartz et al. 2003) is used to generate local alignments that are then combined to create a nonoverlapping pairwise alignment, allowing for the possibility of local inversions. The human genome is scanned to determine regions that have alignments to all the non-human species. Multiple sequence alignments are constructed using ClustalW (Larkin et al. 2007). In step 3, alignments are scanned to determine divergent sites, after which aggressive filters are applied (see Methods).








