
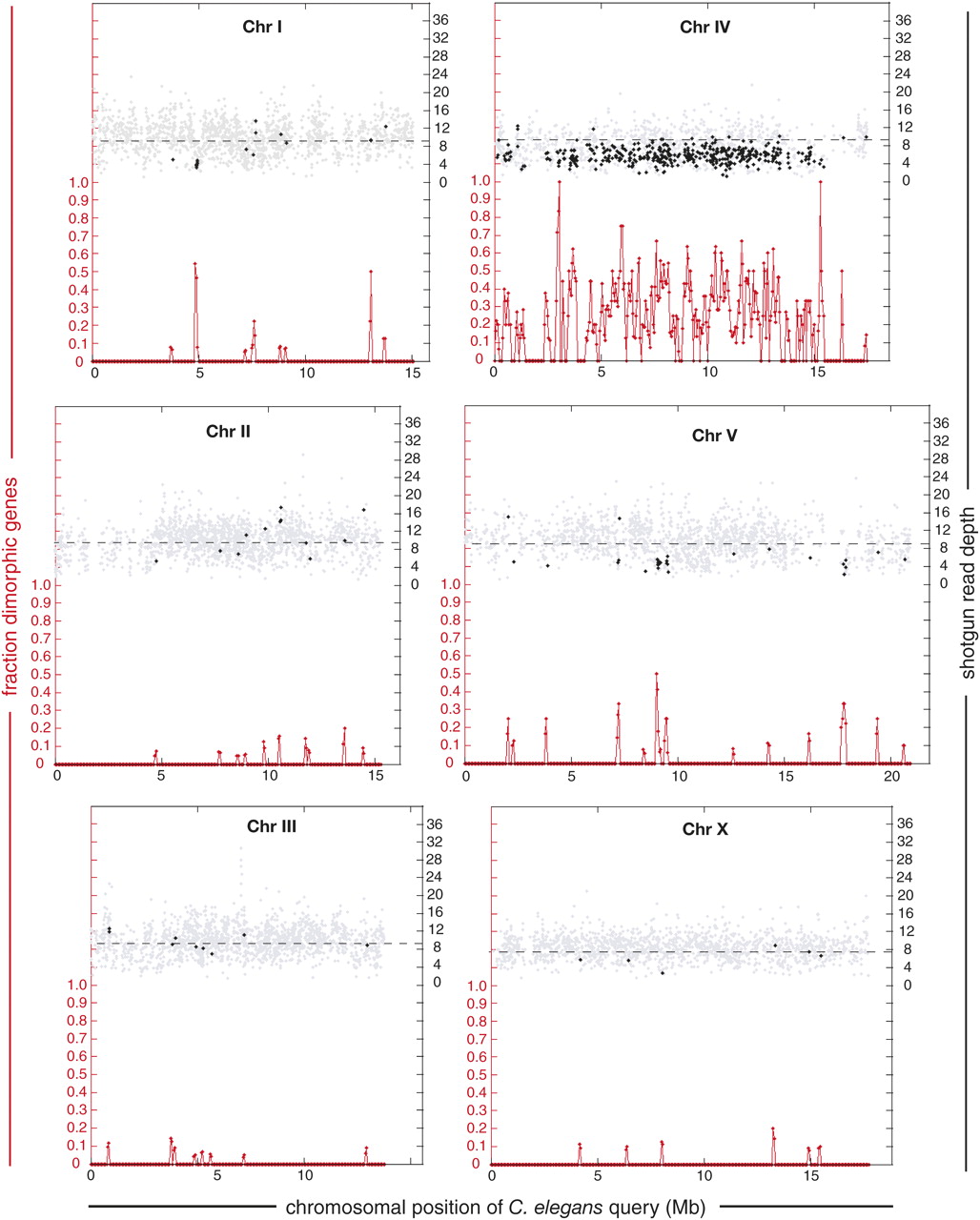
Gene-based, genome-wide survey for heterozygosity in the preliminary C. remanei assembly Cr01. 10,322 single-copy C. elegans genes were used to query the assembly. The fraction of total queries that identified two distinct yet highly similar gene predictions within a 100-kb sliding window (with 50-kb steps) along the C. elegans chromosome is plotted at the bottom of each panel. Left scales (red) refer only to these values. The upper portion of each panel depicts the WGS read depth for queries that have an apparent singleton C. remanei homolog (gray diamonds), and the mean depth for doublet homologs (black diamonds). Right scales (black) refer only to these values. The small proportion of queries identifying more than two variants are not shown in the depth analysis. Regions in which doublet homologs occur in clusters and have consistently low mean read depth are inferred to be heterozygous. By this criterion, regions of the C. remanei genome that are syntenic with C. elegans LGI at 5 Mb, LGV at 9 and 18 Mb, and nearly all of LGIV are heterozygous. The mean WGS read depth for the singleton homologs in each query chromosome is plotted with a dashed line. The singleton read depth for chromosome X (8.23×) lies between the genome-wide 9.2× and the 6.9× expected for equal sex ratio, likely due to the substantially smaller size and genome copy number of males relative to females.








