
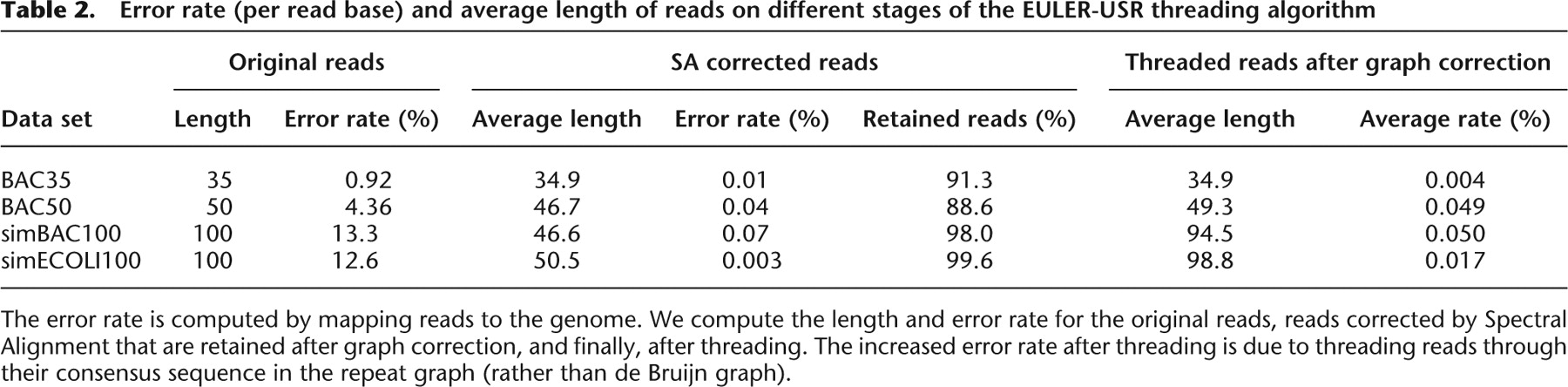
Table 2.
Error rate (per read base) and average length of reads on different stages of the EULER-USR threading algorithm
Click on table to view larger version.
-
The error rate is computed by mapping reads to the genome. We compute the length and error rate for the original reads, reads corrected by Spectral Alignment that are retained after graph correction, and finally, after threading. The increased error rate after threading is due to threading reads through their consensus sequence in the repeat graph (rather than de Bruijn graph).








