
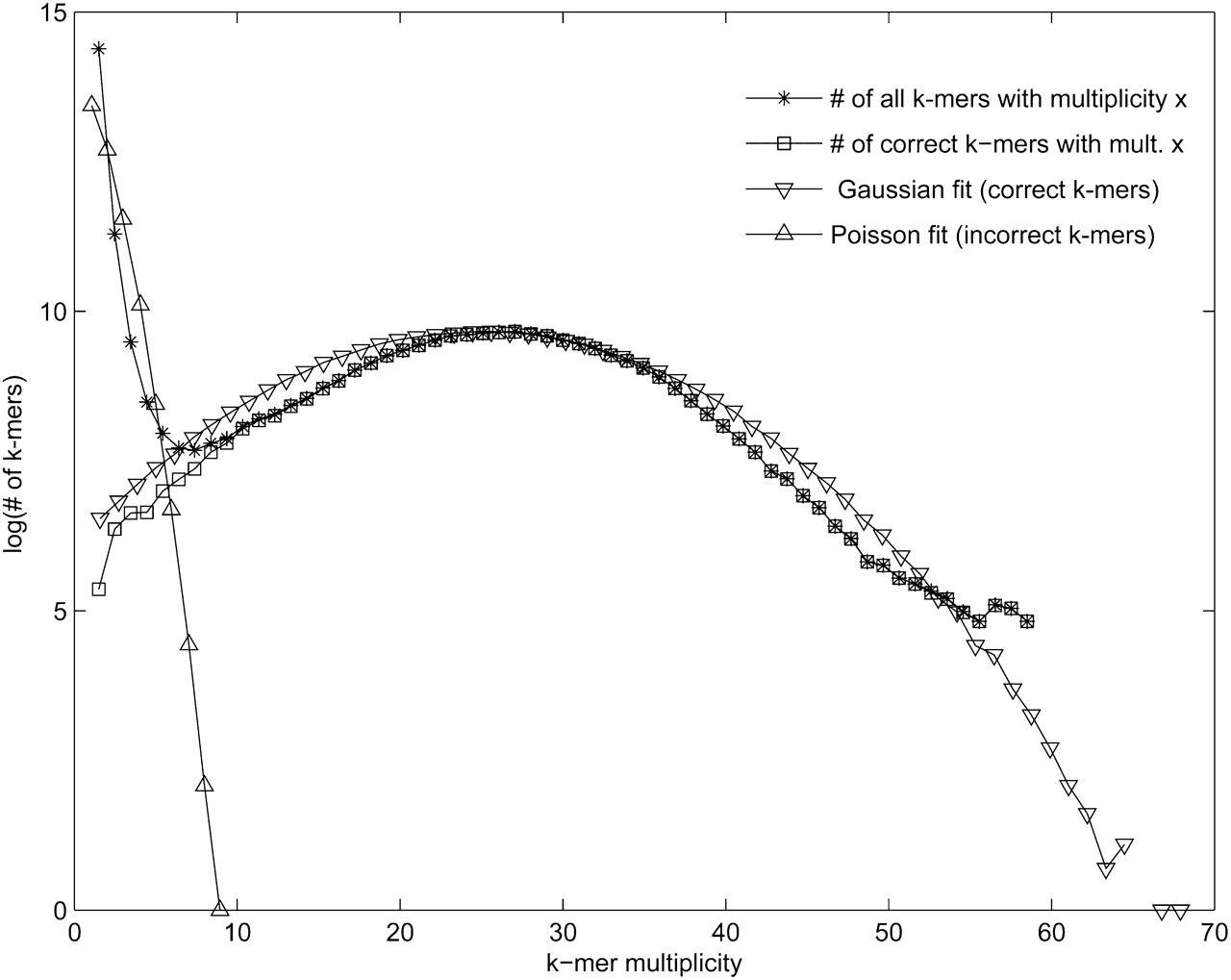
Choosing the multiplicity threshold for error correction. All k-mers appearing in the reads are classified as correct if they appear in the genome, and incorrect otherwise. For a multiplicity x, let correct(x)/incorrect(x) be the number of correct/incorrect k-mers with multiplicity x (the plots are shown for 50-base long Illumina reads from a human BAC and k = 20). As expected, most high-multiplicity k-mers are correct and most low-multiplicity k-mers are incorrect. A Poisson/Gaussian mixture model was fit to the distribution of all k-mer multiplicities in order to model the process of generating incorrect (Poisson) and correct k-mers (Gaussian). To show the fit of the model, the k-mer multiplicities were generated according to the estimated parameters λ = 0.95, μ = 25, and σ = 9.38 with a mixing parameter w = 0.95. One may find the multiplicity m with good separation between correct and incorrect k-mers by estimating the first local minimum from the distribution of k-mer counts or the minimum of the sum of the probabilities of the mixture model, a more smooth distribution. For multiplicity threshold m = 5, only 0.6% of correct 20-mers have multiplicity <5 and only 0.3% of incorrect 20-mers have multiplicity ≥5.








