
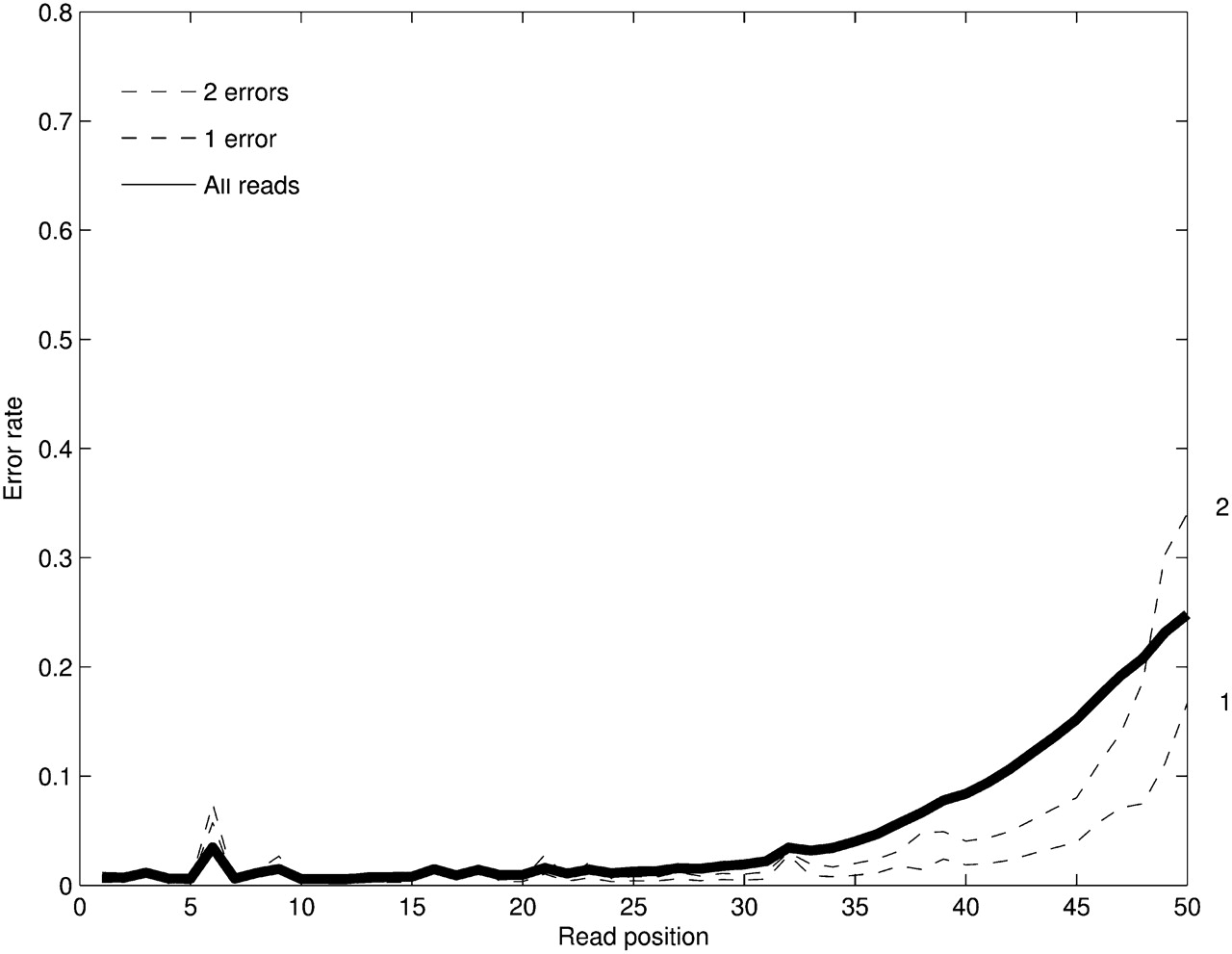
Figure 1.
The positional profile of base-calling errors for Illumina reads for 2 million 50-nt-long reads from a human BAC. The error rate across reads is shown (solid line) along with the error rate for reads with a fixed number of errors. The erroneous nucleotides in each read are detected by mapping the read to the reference genome. The high error rate in position 6 is due to the bias in our particular data set rather than a systematic problem with the Illumina technology.








