
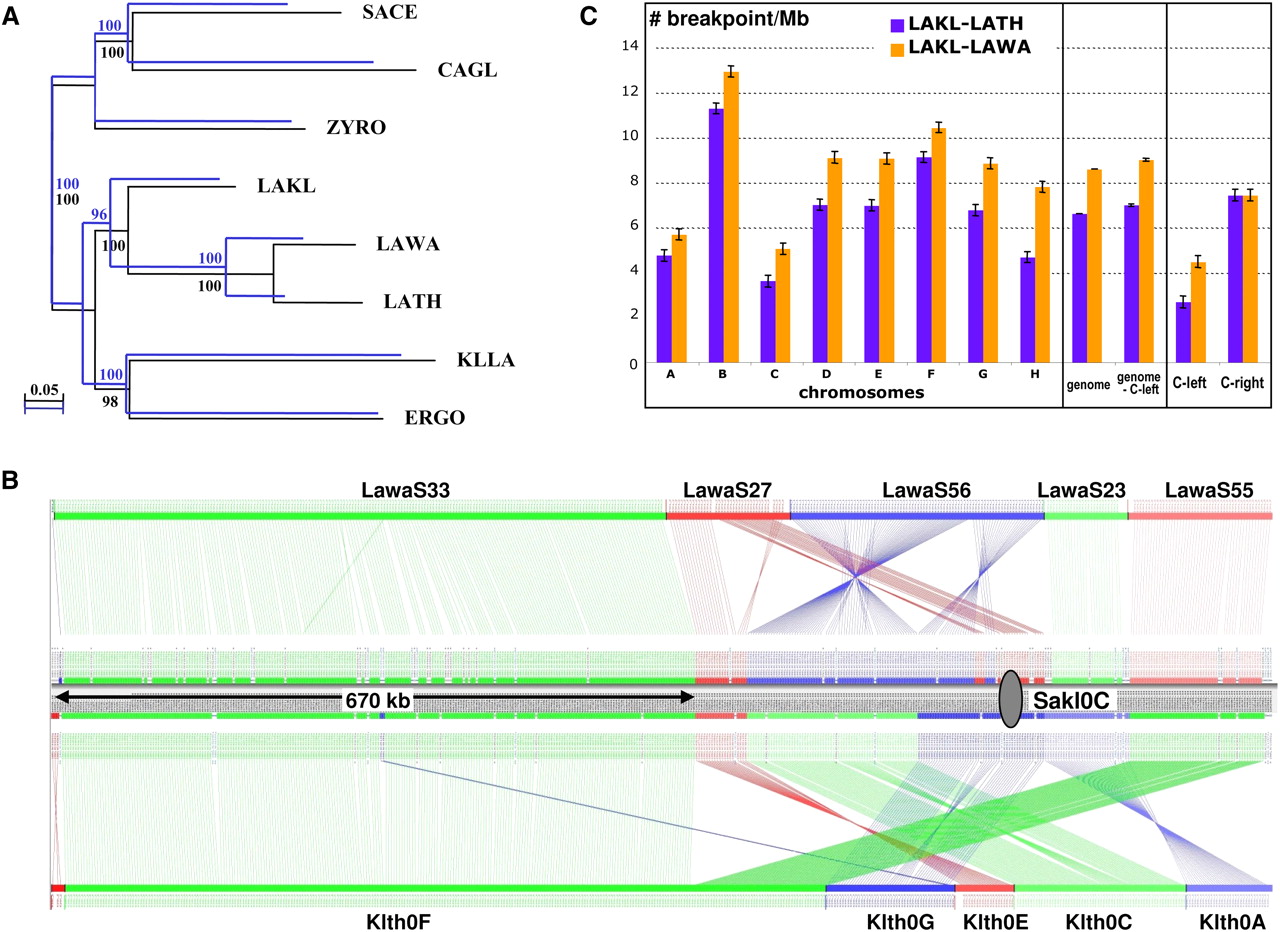
Phylogeny and synteny analyses of L. kluyveri. (A) Two phylogenetic trees were constructed from alignments of universally conserved proteins encoded by C-left (blue; 19 families, 4631 residues) or in the rest of the genome (black; 17 families, 6688 residues) using PhyML, a maximum likelihood method. Species belonging to the Lachancea clade, L. thermotolerans (LATH) and L. waltii (LAWA), were used, as well as more distantly related species such as Kluyveromyces lactis (KLLA), E. gossypii (ERGO), Zygosaccharomyces rouxii (ZYRO), Candida glabrata (CAGL), and S. cerevisiae (SACE). (B) Conservation of synteny was calculated with the AutoGRAPH algorithm (Derrien et al. 2007) from reciprocal best hits between proteins encoded by C-left of L. kluyveri and the proteomes of L. waltii and L. thermotolerans. Each vertical line links orthologs between chromosome C of L. kluyveri and the genomes of L. waltii (top) and L. thermotolerans (bottom). (Gray oval) Centromere of chromosome C. (C) Density of synteny breakpoints per megabase (ordinate), between the genome/chromosomes of L. kluyveri and L. thermotolerans (blue bars) and L. waltii (orange bars). The error bars represent the 99% confidence intervals.








