
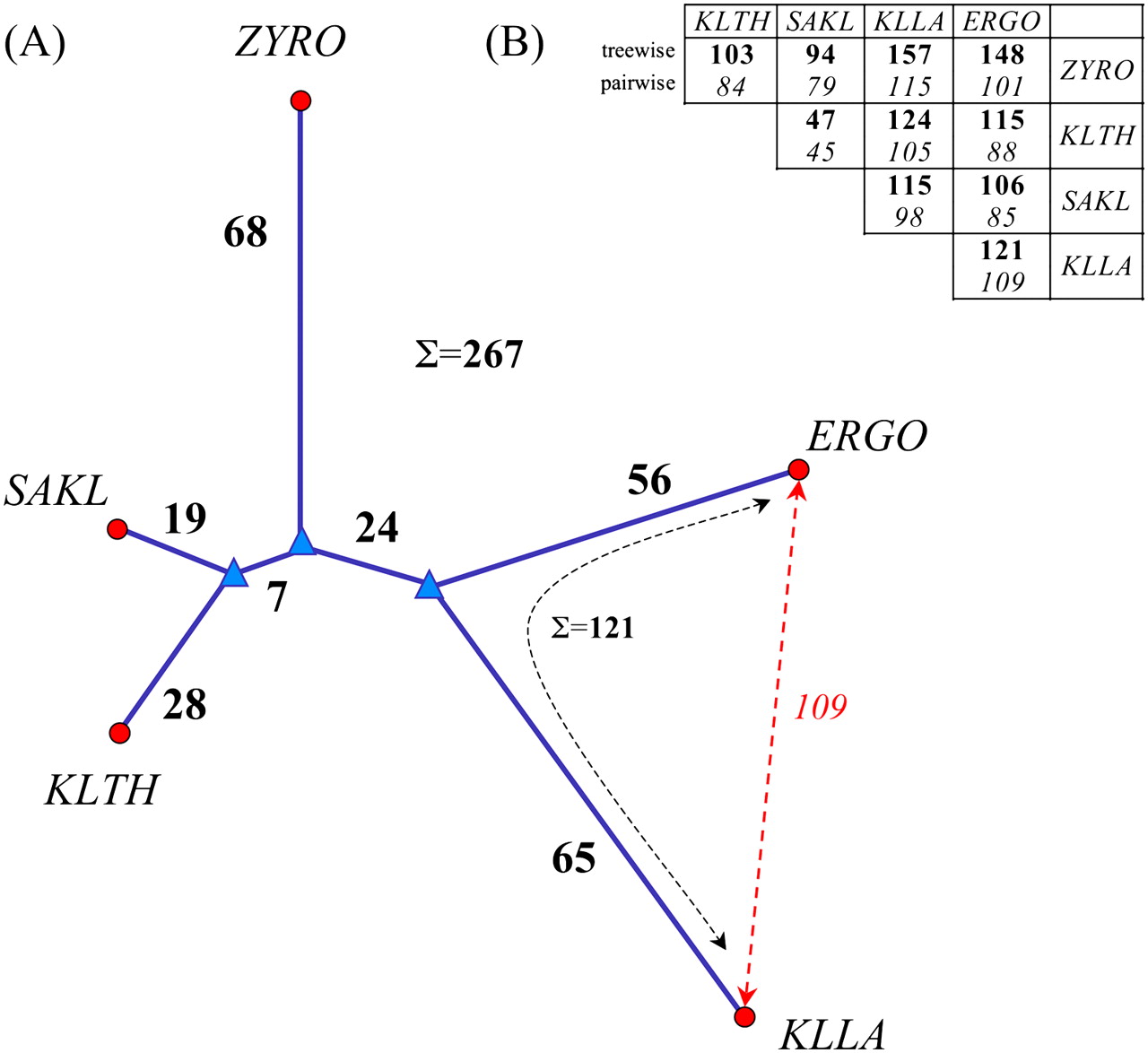
Genome rearrangements. (A) Minimum rearrangement tree computed using the FAUCILS stochastic local search method (Goëffon et al. 2008); (●) contemporary genomes; (▲)inferred ancestral genomes; bold figures show the number of rearrangements in this minimum tree. (B) Contrast between rearrangement distances summing the branches in the tree (in bold, upper row), and pairwise distances (in italics, lower row) computed using the GRIMM method (Tesler 2002). Note that the requirement of going through a common ancestor usually increases treewise distances compared to pairwise distances owing to the triangle inequality, but decreases the sum of distances for the entire tree when all genomes are taken into account. This is illustrated in A for the pair ERGO and KLLA, Σ = 121 > 109, while the overall sum for the tree is 267. The rearrangement tree independantly corroborates the phylogenetic tree calculated using sequence similarity (see Discussion).








