
Abstract
Genome data are increasingly important in the computational identification of novel regulatory non-coding RNAs (ncRNAs). However, most ncRNA gene-finders are either specialized to well-characterized ncRNA gene families or require comparisons of closely related genomes. We developed a method for de novo screening for ncRNA genes with a nucleotide composition that stands out against the background genome based on a partial sum process. We compared the performance when assuming independent and first-order Markov-dependent nucleotides, respectively, and used Karlin-Altschul and Karlin-Dembo statistics to evaluate the significance of hits. We hypothesized that a first-order Markov-dependent process might have better power to detect ncRNA genes since nearest-neighbor models have been shown to be successful in predicting RNA structures. A model based on a first-order partial sum process (analyzing overlapping dinucleotides) had better sensitivity and specificity than a zeroth-order model when applied to the AT-rich genome of the amoeba Dictyostelium discoideum. In this genome, we detected 94% of previously known ncRNA genes (at this sensitivity, the false positive rate was estimated to be 25% in a simulated background). The predictions were further refined by clustering candidate genes according to sequence similarity and/or searching for an ncRNA-associated upstream element. We experimentally verified six out of 10 tested ncRNA gene predictions. We conclude that higher-order models, in combination with other information, are useful for identification of novel ncRNA gene families in single-genome analysis of D. discoideum. Our generalizable approach extends the range of genomic data that can be searched for novel ncRNA genes using well-grounded statistical methods.
Non-coding RNAs (ncRNAs) have long been regarded as the exception to the rule that proteins are the major functional component of the cell. However, the recent discovery of a large number of functional ncRNAs, such as microRNAs (miRNAs), have led to a paradigm shift in which ncRNAs are seen to play a major role in the regulation of gene expression (Aravin and Tuschl 2005).
Computational gene prediction plays an increasingly important, but challenging, role in the identification of new ncRNA genes (Hüttenhofer and Vogel 2006). Although ncRNA gene homologs often contain sequence motifs that are conserved among distantly related organisms, these motifs are generally too short to be used as a sole search criterion. In addition, the structure of an ncRNA is often important for its function but difficult to confidently predict or use for genomic searches (Eddy 2002). Hence, methods for computational identification of ncRNA genes are usually specialized for a particular class of ncRNAs whose characteristics are already well known, for example, tRNAs, snoRNAs, or RNase P RNA (Lowe and Eddy 1997, 1999; Edvardsson et al. 2003; Laslett and Canbäck 2004; Griffiths-Jones et al. 2005; Piccinelli et al. 2005; Kyriakopoulou et al. 2006; Schattner et al. 2006). In contrast, de novo ncRNA gene prediction without a priori knowledge of RNA structure and function is difficult. Most progress in this area relies on the identification of conserved sequences and predicted structures through genome comparisons (Rivas and Eddy 2001; Havgaard et al. 2005; Washietl et al. 2005; Pedersen et al. 2006). Therefore, there is room for improvement of methods to find new ncRNA genes in genomes for which related genomes have not yet been sequenced.
The organism in this study, the social amoeba Dictyostelium discoideum, has an AT-rich (78%) genome of 34 Mb and about 12,500 protein-coding genes (Eichinger et al. 2005). Evolutionarily, D. discoideum is placed between the plant and animal–fungi lineages (Baldauf et al. 2000). D. discoideum live on the forest floor as single cells, where they feed on bacteria. Upon starvation, the cells embark upon a developmental program in which up to 100,000 cells move together and differentiate, forming a multicellular-like organism. Recent experimental and computational analyses have identified a large number of expressed and predicted ncRNA genes in D. discoideum, including genes belonging to known ncRNA gene families as well as unique ncRNA classes that have not previously been described (Aspegren et al. 2004; Kuhlmann et al. 2005; Hinas et al. 2006; Hinas and Söderbom 2007).
Rivas and Eddy (2000) first applied nucleotide compositional contrasts in the average nucleotide composition to identify ncRNA genes within an AT-rich genome. Their work was extended by Klein et al. (2002) and Schattner (2002), who separately used the nucleotide contrast approach to identify 10 and 19 ncRNA candidates, respectively, in the AT-rich (∼69%) genome of Methanococcus jannaschii. In total, 23 unique candidates were identified, and Klein et al. (2002) showed expression from five out of their 10 candidates. Another organism with an extremely AT-rich (80%) genome is the malarial parasite Plasmodium falciparum (Gardner et al. 2002). Its genome was recently searched for ncRNA genes using a compositional contrast approach combined with a search for conserved regions among three closely related Plasmodium species (Upadhyay et al. 2005). This study reported 18 candidates for new ncRNA genes, of which six could be detected by Northern blot analysis (Upadhyay et al. 2005).
The compositional contrast approach to predict ncRNA genes requires methods to locate compositionally deviating segments within a sequence, to measure the statistical significance of the compositional deviations, and to define the boundaries of the deviating segments. Schattner (2002) and Upadhyay et al. (2005) both used a sliding window to segment genome sequences based on average GC content. The sliding-window approach is attractive since it is relatively uncomplicated and large genomes can be rapidly searched. However, it suffers from complementary problems in the lack of a well-grounded theoretical basis for statistics and difficulty in achieving precision in defining boundaries of the segments. Instead of a sliding-window approach, Klein and coworkers used a two-state hidden Markov model (HMM) to identify GC-rich segments (Klein et al. 2002). The HMM provides a much more flexible framework with natural solutions for boundary detection. However, a relatively simple and straightforward HMM approach, as used, for instance, by Klein et al. (2002) carries implicit a priori assumptions about the length distributions and genomic dispersions of ncRNA genes. Also, the statistical significance of candidates obtained from the sliding-window or HMM approaches cannot be calculated analytically but, rather, are computed by parametric simulation.
An alternative approach to segment an input sequence is to transform it into a sequence of scores according to a set of rules. Within this sequence, disjoint segments are determined so that the partial sum of the segment is maximized. Karlin and Altschul have proposed several suitable rules and have studied distributions of maximum segment scores, enabling the calculation of the statistical significance of such segments (Karlin and Altschul 1990; Karlin et al. 1990). This has applications to various problems, most notably sequence comparison, for which it has been implemented in BLAST (Altschul et al. 1990, 1997). It has also been used for other purposes such as identification of transmembrane domains and predicting replication origins in viral genomes (Karlin and Altschul 1990; Chew et al. 2007). In a recent work, Csűrös (2004) developed an algorithm for optimally combining disjoint segments and used the Karlin-Altschul theory for controlling the statistical significance of combined segments.
It is well known that structural features of ncRNA often are important for the function of ncRNAs (Eddy 2002). The best algorithms in use today for RNA secondary structure prediction by free energy minimization rely on the empirical nearest-neighbor model (for review, see Mathews 2006). Overlapping dinucleotide frequencies have proven important to statistically model RNA sequences (Workman and Krogh 1999; Clote et al. 2005), at least in part because the nearest-neighbor model includes base-pair stacking increment parameters within stems (Xia et al. 1998). We therefore reasoned that the compositional contrast approach might be more powerful for the ncRNA gene-finding application if it were generalized to consider overlapping dinucleotides. Fortunately, Karlin and Dembo (1992) derived results that generalize Karlin-Altschul statistics to Markov-dependent sequences. However, to our knowledge, their results have not been applied before to this or any other biological problem.
In this study, we implemented a partial sum process using empirical log-likelihood ratio scoring schemes and Karlin-Altschul and Karlin-Dembo statistics and applied it to de novo ncRNA gene prediction in the AT-rich genome of the protist D. discoideum. With this method, we recovered 94% of previously identified ncRNA genes. The predictions were subsequently filtered using biologically significant criteria such as the repeated occurrence of similar sequences within the D. discoideum genome or the nearby presence of a Dictyostelium upstream sequence element (DUSE) previously implicated to be associated with ncRNA genes (Aspegren et al. 2004; Hinas et al. 2006; Hinas and Söderbom 2007). Finally, we verified expression of six out of 10 novel ncRNA genes or gene families by Northern blot analysis. We believe that our improved contrast approach to de novo single-genome prediction of ncRNA genes will be a useful search tool also for other genomes where a sufficient compositional contrast exists and will be particularly powerful in combination with other search criteria.
Results
Nucleotide and dinucleotide composition of ncRNA genes and the background search space in D. discoideum
Genes expressing ncRNAs often have higher GC content than their surrounding DNA context, especially in organisms with AT-rich genomes (Rivas and Eddy 2000). This observation encouraged us to analyze the nucleotide compositions of ncRNA genes known or confidently predicted to be expressed in D. discoideum (Table 1; Hinas and Söderbom 2007), including 379 tRNA genes that we predicted using the ARAGORN tRNA prediction software (Laslett and Canbäck 2004). The average GC content of the ncRNA genes ranges from ∼33% for the RNase P RNA gene up to ∼51% for the tRNA genes (Table 1). The GC content of the ncRNA genes was considerably higher than the genomic average of 22% (Eichinger et al. 2005). Furthermore, the vast majority of these were found in nonrepetitive intergenic regions where the average GC content was only ∼14%. Thus, the contrast in average GC content between the ncRNA genes and the nonrepetitive, intergenic or intronic genome of D. discoideum (the “background genome”; see Methods) is approximately two- to 3.5-fold.
Known D. discoideum ncRNAs
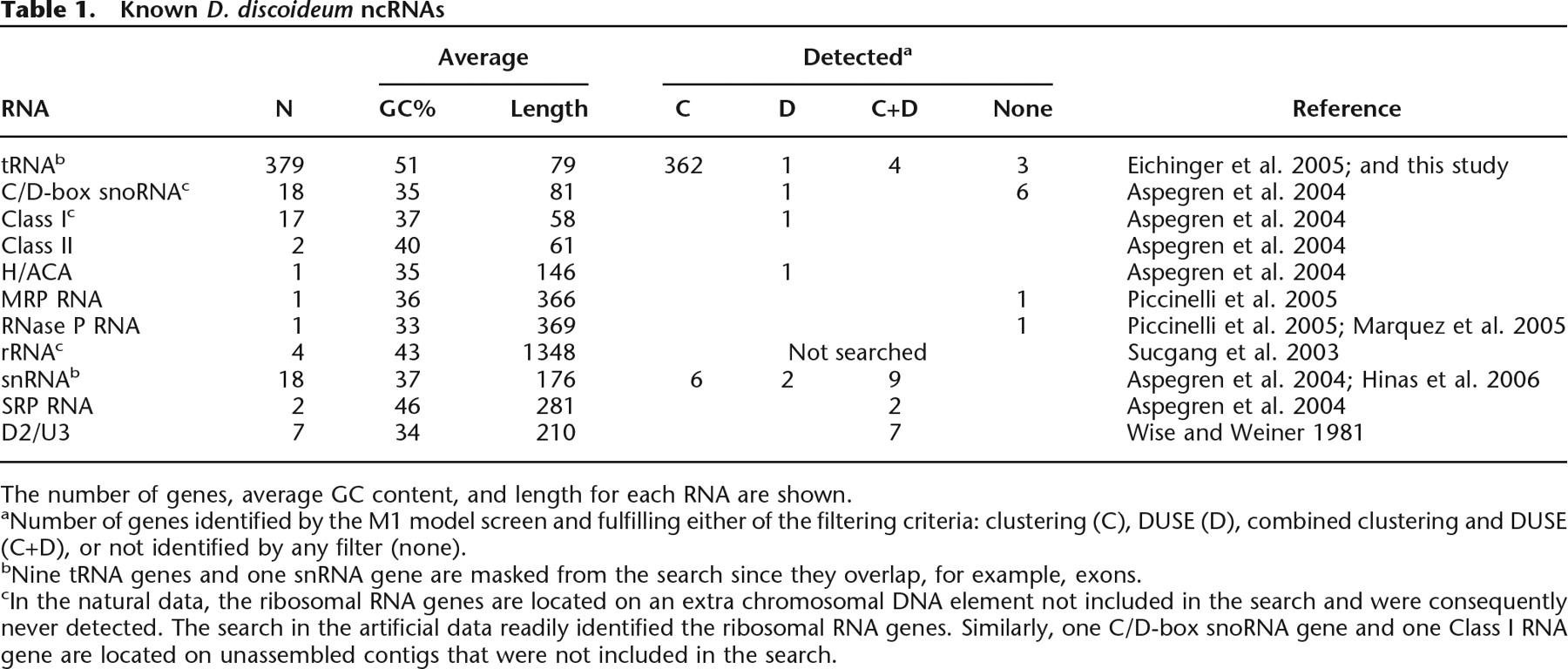
The number of genes, average GC content, and length for each RNA are shown.
aNumber of genes identified by the M1 model screen and fulfilling either of the filtering criteria: clustering (C), DUSE (D), combined clustering and DUSE (C+D), or not identified by any filter (none).
bNine tRNA genes and one snRNA gene are masked from the search since they overlap, for example, exons.
cIn the natural data, the ribosomal RNA genes are located on an extra chromosomal DNA element not included in the search and were consequently never detected. The search in the artificial data readily identified the ribosomal RNA genes. Similarly, one C/D-box snoRNA gene and one Class I RNA gene are located on unassembled contigs that were not included in the search.
In order to investigate the possibility of predicting the orientation of the candidate ncRNA genes, we analyzed the strand symmetry for the background genome and the known ncRNA genes. While the background genome is nearly strand-symmetric, there is some asymmetry in ncRNA strands (Supplemental Data S1 and S2). However, when this was applied to determine the coding strand for the candidate ncRNA genes, the predictions were not reliable (data not shown). Hence, we did not discriminate between the strands during our search.
Considering dinucleotides, G-tests of independence (Zar 1999) strongly reject independence of consecutive nucleotides (p ≪ 0.001, not surprisingly given the large sample sizes). Furthermore, and in agreement with previous studies (Karlin and Burge 1995; Karlin et al. 1998), the background had stronger departures from those expected by independent mononucleotides with strong tendencies toward repeats (i.e., “CC” and “GG” are more than 2.4 times more prevalent than expected). For D. discoideum ncRNA genes, TC is most overrepresented among dinucleotides and exceeds the expected frequency 1.3-fold, while AC is most underrepresented at ∼71% of expectation. Comparing conditional dinucleotide probabilities by ratios of those in target to background revealed substantial contrasts, such as a greater than sixfold higher conditional probability of G following an A in targets. This indicates that using higher-order Markov-dependent partial sum processes could yield better results in ncRNA gene finding as detailed below. Based on these results, we were encouraged to pursue a screen for ncRNA genes based on contrasts of nucleotide and dinucleotide contents.
Strategy to search for ncRNA genes based on deviations in nucleotide and dinucleotide compositions
In order to search for new ncRNA genes in available genome sequences, we implemented a segmentation approach based on maximal partial sums in a sequence of scores. To evaluate the statistical significance of segments, we used Karlin-Altschul statistics for independent nucleotides (“the M0 model”) and the Karlin-Dembo extension for nucleotides with first-order Markov dependence (“the M1 model”) (Karlin et al. 1990; Karlin and Dembo 1992). The only parameters required are the rules for scoring a nucleotide sequence and the probabilities of assigning each score. Karlin and Altschul (1990) proposed a scoring scheme using log-likelihood ratios of frequencies of word occurrence events in a “target” distribution over those in a “null” or “background” distribution. We derived such a scheme by calculating target nucleotide word frequencies from a nonredundant set of known or inferred D. discoideum ncRNA genes (frequencies were calculated from both the forward and reverse complement sequence). Background frequencies were derived from the D. discoideum genome, where complex repeats, exons, and known or inferred ncRNA genes had been removed (see Methods). The relative entropies of the scoring matrices in the two models indicate that conditional dinucleotides have more than three times the information for detecting target ncRNA genes than mononucleotides (for M0, the entropy is 0.19 bits per nucleotide; and for M1, the entropy is 0.68 bits per nucleotide). If nucleotides were independent, the expected information increase of the conditional dinucleotide matrix would be merely twice that of the mononucleotide matrix.
Performance evaluation
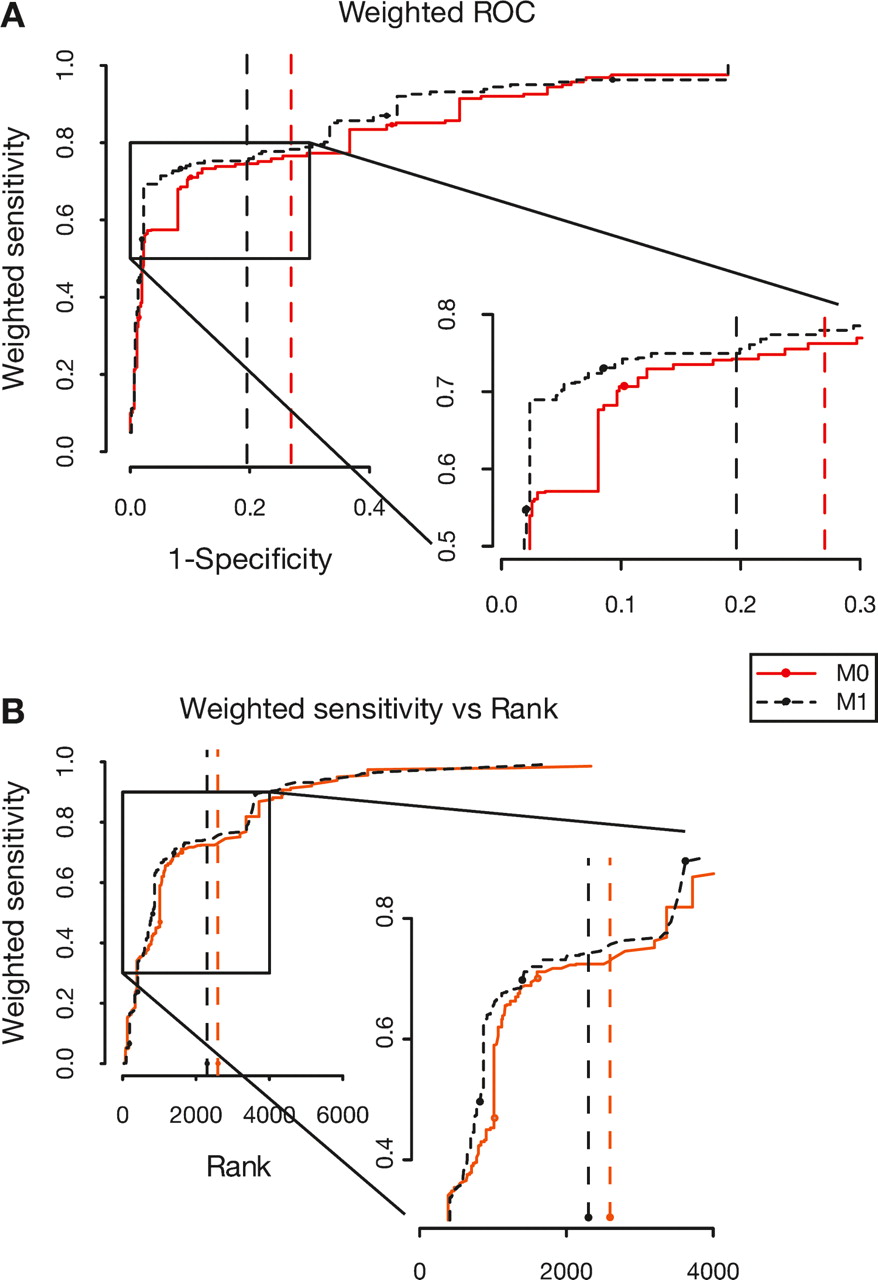
To compare the performances of the M0 and M1 models, we generated a simulated search space of the same size as the D. discoideum genome using a Markov chain of fifth order (i.e., the probability of generating each individual nucleotide depends on the previous five nucleotides). We estimated parameters for the Markov chain from the repeat masked D. discoideum intergenic regions as detailed in Methods. The background was masked and known or confidently predicted ncRNA genes were inserted at the same locations as in the natural data. We could then compare the performance of the M0 and M1 models by analyzing the sensitivity and specificity in rediscovering the inserted ncRNA genes. We scored each strand independently but considered a prediction as a true positive if it overlapped a known ncRNA, regardless of the strand and the length of the overlap. Predictions that overlapped each other but on opposite strands were combined into one prediction using the outermost endpoints. To evaluate the performance, we calculated the area under the curve (AUC) when sensitivity was plotted against the false positive rate (1-specificity) (Fig. 1A, ROC-plot). The sensitivity was weighted according to the ncRNA family as described in Methods. With AUC 0.84 and 0.87 for the M0 and M1 models, respectively, we conclude that the M1 model performs better on the artificial data. We performed a twofold cross-validation (see Methods). When using the artificial background data, the mean of the AUC for M0 and M1 was 0.76 and 0.80, respectively (standard deviations 0.04 and 0.02, respectively). When using background data sampled from the D. discoideum genome, the mean AUC was 0.21 and 0.22 for M0 and M1, respectively (standard deviation 0.02 and 0.005).
Performance of the M0 and M1 models. (Solid red line and red circles) Performance of M0 model. (Dotted black line and black circles) Performance of M1 model. (Dashed lines) The E = 0.1 threshold for the M0 (red line) and M1 (black line) models, respectively. (A) An artificial background was generated (see Methods). The performance of the M0 and M1 models was measured in terms of sensitivity and specificity. The sensitivity was weighted so that each ncRNA family has an equal contribution to the total sensitivity. The weighted sensitivity is plotted against 1-specificity. (B) Performance on real data. The weighted sensitivity in detecting known or confidently predicted ncRNAs in the D. discoideum genome is plotted against the rank of the corresponding hits.
Search for novel ncRNA genes in D. discoideum
Approximately 12 Mb of the D. discoideum genome that remained after masking of exons and repeats was searched in both directions using both the M0 and M1 models (see Methods). Figure 1B shows the sensitivity weighted by ncRNA family plotted against the ranking of corresponding hits for both models. It can be seen that the models perform similarly but the M1 model has a higher specificity as the number of hits increases. This prompted us to proceed with the M1 model predictions to identify candidates that could potentially correspond to novel ncRNA genes, and we found an E-value cutoff of 0.1 (Fig. 1B, dashed, vertical line) to be a good compromise between sensitivity and the number of hits. The search strategy is outlined in Figure 2.
Flowchart and clustering of sequences. The six chromosomes of the D. discoideum where exons and complex repeats had been masked were searched for high-scoring segments with an expect value of at most 0.1. Overlapping high-scoring segments residing on opposite strands were combined. The predictions were filtered for presence in multi-copy families and/or the occurrence of an upstream sequence element (DUSE) within 150 nt flanking the prediction. Piecharts represent the number and character of sequences that were used for the operation. The size of the piechart is proportional to the total number of sequences. (Yellow and the associated numbers) Novel predictions; (blue and associated numbers) known or confidently predicted ncRNA genes. After combining the strands, the number of known or confidently predicted ncRNA genes increased since genes with an overlapping prediction on the template strand count as detected after the combination step.
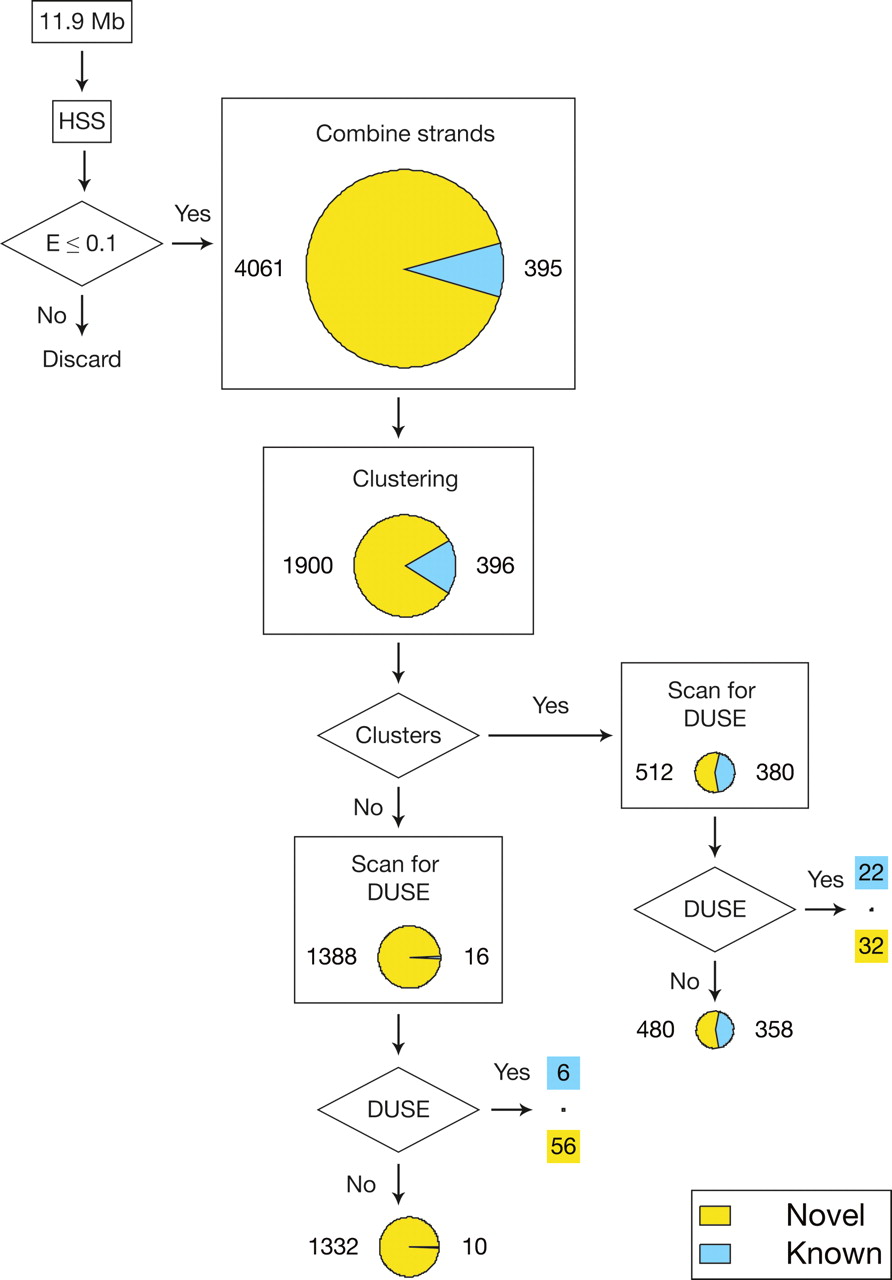
This search strategy resulted in 2296 candidate ncRNA gene-containing regions. We refer to these as drp for “Dictyostelium ncRNA predictions.” Of the 2296 predicted regions, 396 overlapped known or confidently predicted D. discoideum ncRNA genes. The shortest overlap between a prediction and a known ncRNA gene observed was 35 nt or 45% of the length of the ncRNA. Ten predictions each overlap two closely located tRNA genes, and one prediction contains two C/D-box snoRNAs. Hence, a total of 94% or 407 of the 435 known or confidently predicted ncRNA genes (not overlapped by repeats or protein-coding genes) were successfully predicted by our method (Table 1), including 370 of 379 tRNAs, 17 of 18 snRNAs, the H/ACA-box snoRNA, RNase P and MRP RNAs, SRP RNAs, and seven of 18 C/D-box snoRNAs. In the artificial background, the same sensitivity is reached at a false positive rate of ∼25%, although this number is most likely an underestimation because of the ideal nature of this data set.
In order to identify the predicted regions most likely to contain new ncRNA genes, we applied different filtering procedures (Fig. 2). The first filter was based on the observation that known ncRNA genes often occur in multigene families, that is, similar or identical RNA genes are often present in multiple copies throughout the genome. This organization also applies to the ncRNA genes in D. discoideum (Hinas and Söderbom 2007). Therefore, the candidates were clustered according to sequence similarity. From here on, we will use “cluster” to describe two or more sequences that appear related by sequence similarity (not implying location in the genome). We use “family” to refer to clusters that we believe represent homologous ncRNA genes. Our analysis generated 131 distinct clusters with two to 159 members (Supplemental Fig. S1). In total, 892 of the 2296 putative ncRNAs could be clustered by sequence similarity. More than half of the clusters contained only two members, and roughly one-third of the two-member clusters (25 of 73) can be explained by the 750-kb genomic duplication previously described (Eichinger et al. 2005). Known D. discoideum ncRNAs were present in 21 of the identified clusters (Table 1; Supplemental Fig. S1).
As a second filter, we searched for the presence of a ncRNA-gene-associated upstream sequence element within 150 nt flanking the 2296 predicted gene-containing regions. This 8-nt putative promoter element, DUSE, is located ∼63 nt upstream of the transcription start site of the majority of the recently identified and expressed ncRNA genes in D. discoideum (Aspegren et al. 2004; Hinas et al. 2006; Hinas and Söderbom 2007). The DUSE sequence was found in association with 116 predicted ncRNA genes. Of these, 28 correspond to genes already known or confidently predicted to express ncRNAs. We also combined the two filters to search for sequences within clusters that are associated with DUSE, which resulted in 54 members dispersed in 25 clusters. Nine of these clusters contain known or confidently predicted ncRNA genes.
Genomic distribution
The predictions are in general evenly distributed across the genome, with on average one prediction every 15 kb. The known or confidently predicted ncRNAs occur on average once every 80 kb, but this number varies substantially between chromosomes with Chromosome 6 having the highest ncRNA density of one every 40 kb and Chromosomes 4 and 5 the lowest with one every 120 kb. The authors and others have previously reported that ncRNA genes in D. discoideum frequently occur in pairs or triplets of highly similar copies (Aspegren et al. 2004; Eichinger et al. 2005; Hinas et al. 2006). These copies are located in all possible orientations, that is, tandem, divergent, and convergent, usually within 25 kb of each other but often even closer; for example, some of the snRNA genes are separated only by a few hundred base pairs. Even though the mechanism is unclear, this organization suggests recent duplication events (Eichinger et al. 2005). We found that 34 of our identified clusters of candidate ncRNA genes show a similar organization in the genome; that is, two members are closer than 25 kb apart. In addition, eight of the 12 members of cluster drf9 (drf for “Dictyostelium ncRNA family”) seem to colocalize with ncRNA genes belonging to the Class I RNA genes, a family of short, abundantly expressed ncRNAs only known in D. discoideum (see Supplemental Table S1; Aspegren et al. 2004; Hinas and Söderbom 2007). Despite the close association, it seems unlikely that these genes are co-transcribed since both the Class I genes and the drf9 genes carry their own upstream DUSE sequence, and in many cases, they are located on opposite strands.
Experimental validation
In order to validate the search algorithm used to screen for ncRNA genes, we investigated gene expression by Northern blot analyses on total RNA isolated from growing D. discoideum cells. For each tested candidate ncRNA gene-containing region, we assayed both strands using strand- and target-specific oligonucleotide probes. We chose to analyze the expression of a set of randomly selected candidate RNA genes that could be clustered together by sequence similarity. The probes were designed to recognize the maximum number of members within the same cluster; that is, they were complementary to the most conserved regions.
For the expressed family drf17 (see below), the hybridization oligonucleotide matched five additional sites in the genome. However, upon closer inspection, these sequences were found to be similar to drf17 but did not meet the significance threshold of the search algorithm. Similarly, the probe for drf9 (see below) matched one additional site in the genome, which was positioned within a predicted candidate. Owing to the stringency of the clustering algorithm, the candidate failed to cluster together with drf9.
The search generated families containing sequences of variable length. In the majority of cases, the member sequences within each family have a core motif where most of the nucleotides align (target for the hybridization probes). However, other motifs were sometimes shifted in relation to each other owing to insertions and gaps of nonsimilar sequences.
Eight of the 131 identified clusters were analyzed for expression, and four of these were transcribed into small RNAs (drf115, drf17, drf22, and drf9) (Fig. 3; Table 2). The putative multigene family drf27 yielded signals indicating RNAs substantially longer than the predictions and expressed from both strands, while drf15 showed a hybridization signal of ∼160 nt from one strand. However, the signal disappeared at high stringency washes. Hence, these two families were considered to be false positives. The additional two predicted multigene families, drf32 and drf11, failed to give rise to any detectable hybridization signals.
Candidate ncRNAs analyzed for expression
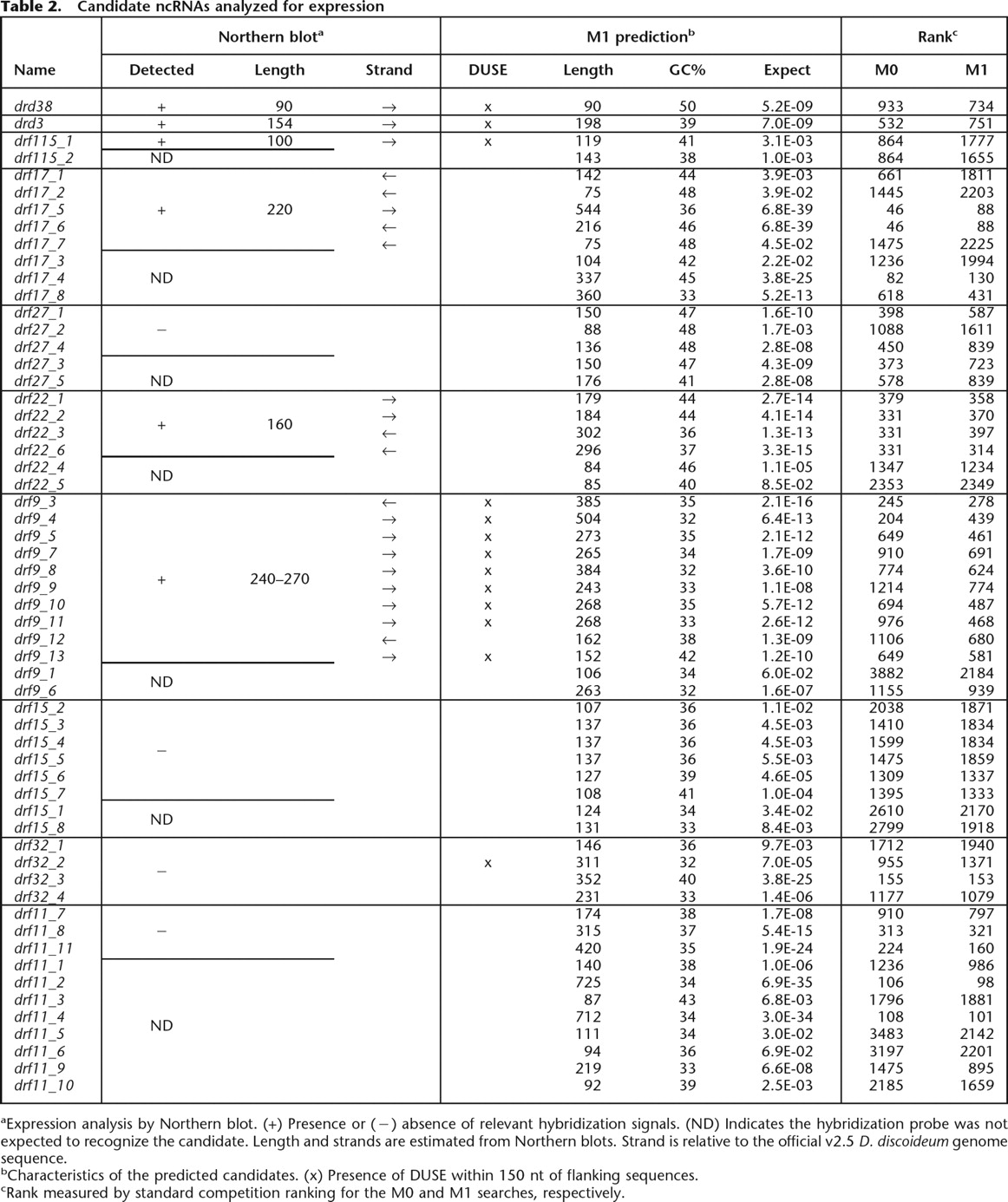
aExpression analysis by Northern blot. (+) Presence or (−) absence of relevant hybridization signals. (ND) Indicates the hybridization probe was not expected to recognize the candidate. Length and strands are estimated from Northern blots. Strand is relative to the official v2.5 D. discoideum genome sequence.
bCharacteristics of the predicted candidates. (x) Presence of DUSE within 150 nt of flanking sequences.
cRank measured by standard competition ranking for the M0 and M1 searches, respectively.
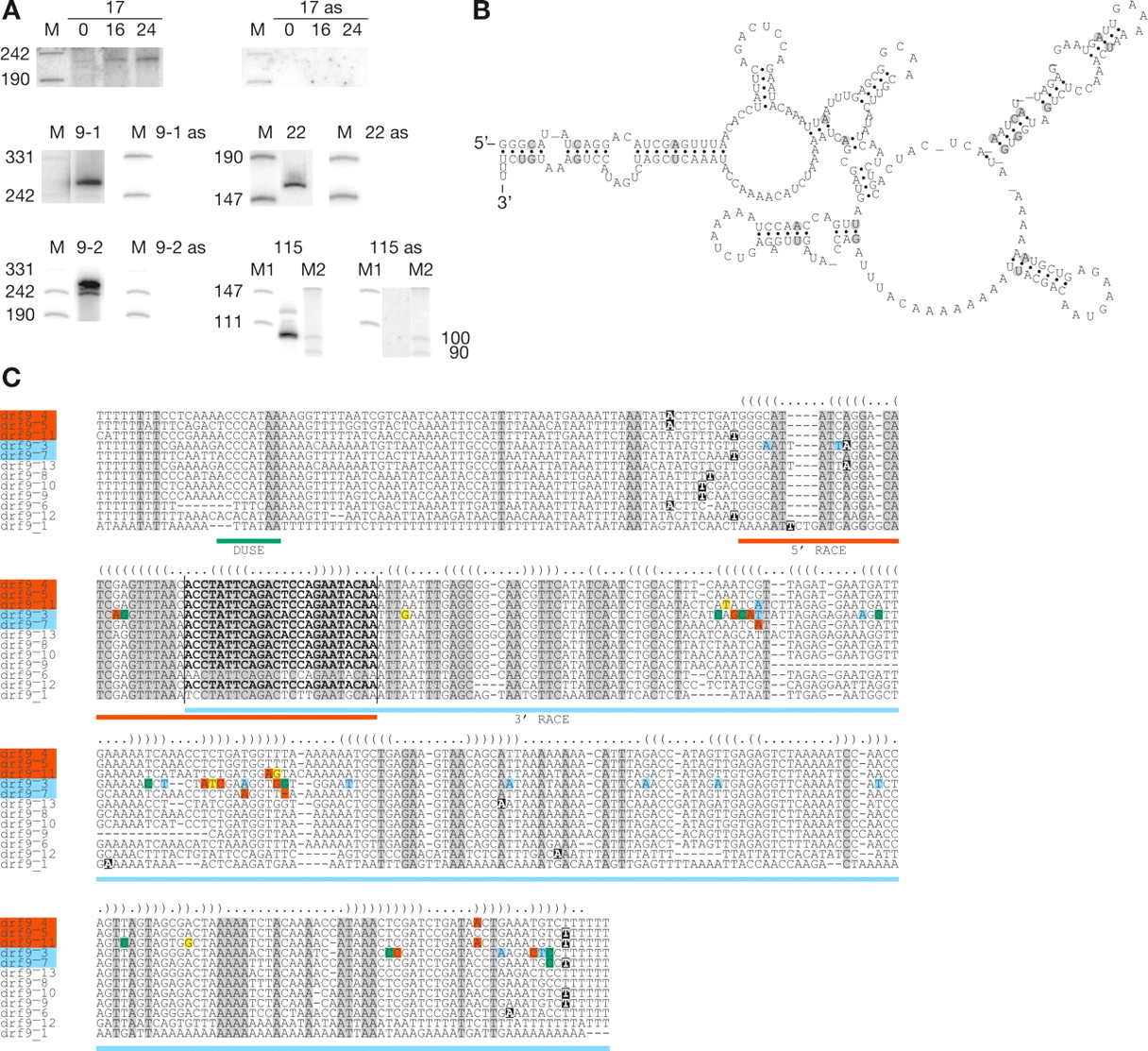
Analysis of expressed ncRNA families. (A) Northern blot analysis of families hybridized with sense and anti-sense (as) probes. drf17 was analyzed for developmental regulation on RNA extracted at 0 h (growing cells), 16 h, and 24 h. Two slightly different probes were used in the analysis of drf9 (9-1 and 9-2). (B) Consensus secondary structure, predicted by RNAalifold, of the five members matching sequences determined by RACE analysis. Compensatory base pair changes are circled. (C) Alignment of the drf9 member sequences including upstream and downstream flanking sequences. (Green box beneath sequences) Location of the DUSE sequence present in nine sequences. (Red and blue boxes beneath sequences) Sequences determined by 5′- and 3′-RACE analysis, respectively. Candidate gene names labeled with red and blue boxes indicate members matching the sequences determined by RACE analysis. (Bold letters between the vertical bars) Nucleotides complementary to the hybridization probes. (Black boxes in the alignment) Start and end point nucleotides of predictions. The predictions for drf9_3, drf9_4, and drf9_8 extend beyond the alignment for an additional 124, 226, and 111 nucleotides, respectively. (Dot-bracket notation above the alignment) The Alifold consensus structure. The notation has been slightly adapted to accommodate the full family alignment. Compensatory base pair changes in the RACE-determined sequences are indicated: (green box) a change to a GC base pair; (blue box) a change to a AT base pair; (yellow box) a change to a GT base pair; (red box) a change that disrupts base-pairing.
Properties of novel expressed ncRNA multigene families
The family drf17 contains eight similar member sequences, ∼220 nt in length according to Northern blot results (Fig. 3A). One of the predictions spans two highly similar copies located on opposite strands. Hence, each copy was treated as an individual candidate (drf17_5 and drf17_6) (Table 2). We could detect only a faint signal by Northern blot analysis on RNA extracted from growing D. discoideum cells. However, we readily detected hybridization signals when we investigated expression during development (i.e., 16-h slugs and 24-h fruiting bodies) (Fig. 3A). The developmental expression was only analyzed for this family. The multigene family drf22 is made up of six sequences, and the major signal detected on Northern blot appears to be ∼160 nt (Fig. 3A). In addition, a heterogeneous population of longer RNAs possibly derived from the different members of this family was visible. An alternative explanation is that the transcripts from this family are of the same size but subjected to polyadenylation as reported for D. discoideum spliceosomal RNAs (snRNAs) (Hinas et al. 2006).
The family drf115 consists of two sequences located merely 80 bp apart. The member recognized by the hybridization probe has a DUSE motif located 65 nt upstream of the predicted start site. Northern blot detects a major ∼100-nt RNA transcript. In addition, a less strong hybridization signal is visible corresponding to an RNA sized ∼130 nt (Fig. 3A).
The family drf9 contains 12 member sequences (Fig. 3C; Table 2) and was subjected to a more detailed analysis. Two slightly different oligonucleotide probes were used to verify expression (Fig. 3A, 9-1 and 9-2). Hybridization signals indicated expressed RNA of ∼240–270 nt. In order to determine the 5′- and 3′- ends of the RNA, drf9 RNA was subjected to Rapid Amplification of cDNA Ends (RACE) analyses (data not shown). The sequence of the 5′ RACE product (only one sequenced) could be derived from three members of drf9 with identical sequences to the determined 5′ end (Fig. 3C). In addition, two sequenced 3′ RACE products determined the 3′ ends as well as most of the full-length RNA sequence of two additional member RNAs (Fig. 3C). Hence, three different RNAs from within this family have been directly verified. The sizes of these RNAs are estimated to be 265 and 267 nt by 3′ end determination and 265–268 nt by the 5′ RACE. These estimations are based on the following observations:
The RNA sequences are similar;
the 5′ and 3′ ends are positioned in short stretches of fully conserved regions;
the major size as determined by Northern blot analysis is ∼270 nt (one of the hybridization primers was also used for 5′ RACE);
the presence of a conserved sequence element (DUSE) predicted/verified to be located 63 nt in front of nine of the 12 members and preceding all the RNAs where the 5′ and 3′ ends have been confirmed;
the genomic region downstream from the experimentally verified 3′ ends consists of a stretch of thymidine residues. This run of Ts is conserved among the members of drf9 and may constitute a termination signal. The RNA polymerase III-transcribed U6 snRNA genes in other organisms have been shown to use stretches of Ts as terminators (Will and Lührmann 2001), and the same organization is present in D. discoideum (Hinas et al. 2006).
The secondary structure of drf9 was predicted by the RNAalifold software (Hofacker et al. 2002) using the five sequences that correspond to the RACE-determined members (Fig. 3C). The predicted consensus secondary structure contains several stem structures where compensatory base-pair changes are present, corroborating the structure (Fig. 3B).
In summary, four out of eight of the computationally predicted clusters of small RNA genes were expressed. One of the expressed ncRNA families showed very low expression in growing cells, which increased during development. This indicates that transcription of some of the predicted genes may have escaped detection since the majority of the analyses were performed on RNA from growing cells only. One predicted ncRNA gene family, drf9, was investigated in more detail, and the presence and partial sequences of at least three member RNAs were verified.
Increased stringency by searching for conserved upstream elements
As an alternative approach to improve the quality of the predictions, we searched for the conserved upstream sequence element, DUSE (see above), in association with the ncRNA candidates. Since the exact site of transcription initiation could not reliably be predicted for the majority of the RNAs, a region of 150 nt flanking the predicted genes was investigated. The DUSE element was found to be associated with 116 candidate RNA genes. Of these, two candidate ncRNA genes (notably not belonging to any apparent clusters) were randomly chosen for further analysis, and both genes were demonstrated to be transcribed. The transcripts correspond to the DNA strand containing the DUSE (Fig. 4) and are located in the cytoplasm (data not shown). drd38 (drd for “Dictyostelium ncRNA with DUSE”) was detected as a major transcript of ∼90 nt that was found to correspond to the recently identified selenocysteine tRNA (Fig. 4A; Shrimali et al. 2005). The major RNA derived from drd3 is ∼155 nt, while a less abundant species of ∼180 nt was also detected (Fig. 4C). The secondary structures of the RNAs were predicted by RNAfold (Hofacker et al. 1994) based on the sizes estimated by Northern blot analysis and the assumption that the start of transcription is located 63 nt from the DUSE sequence (Fig. 4). In addition, the majority of the members of drf9 (whose expression was verified; see above), as well as the verified member of drf115, also have an upstream DUSE sequence.
Analysis of two candidates with an upstream DUSE sequence. Northern blot analyses were performed for each candidate in sense and anti-sense (as) directions as indicated. RNA secondary structure predictions were obtained from RNAfold using the sequence length deduced from the Northern blot analyses and assuming a transcription start site located 63 nt downstream from the DUSE. (A) Northern blots and (B) secondary structure predictions for the drd38 and drd3 candidates.
These results show that combining our algorithm for ncRNA gene prediction with searches for the conserved DUSE element generates an additional level of confidence. All six of the analyzed genes (including one member of drf115 and the three individual genes within drf9 for which expression was verified by RACE) that were preceded by a DUSE were transcribed.
Discussion
The options for de novo ncRNA gene prediction using a single genome are limited (Meyer 2007), and there is a need for improved methods. In the protist D. discoideum, we observed that the known ncRNA genes have a substantially higher GC content and markedly different dinucleotide composition from the background genome sequence. This prompted us to develop a computational search strategy to identify compositionally deviating regions that could contain novel ncRNA genes. For this purpose, we implemented a search algorithm based on maximal segment scores among partial sums and the Karlin-Dembo extension for Markov dependence to the Karlin-Altschul theory on the statistical significance. We used a scoring scheme based on nucleotide word frequencies derived from background and target sequences. There are several major advantages with this method, for example, the statistical significance of scores of ncRNA gene candidates can be readily calculated, and there are few nuisance parameters that need to be estimated.
First-order Markov dependence, i.e., a model where the probability of observing a particular nucleotide depends on the identity of the nucleotide at the previous position, is motivated from a biological point of view. Nearest-neighbor interactions are known to be important for RNA structural prediction as a consequence of, for example, stacking interactions (for review, see Turner and Sugimoto 1988). We implemented algorithms based on this theory using both a model that assumes independent nucleotide positions (the M0 model) and a model that assumes nucleotide positions with first-order Markov dependence (the M1 model). Mononucleotide frequencies as well as conditional dinucleotide frequencies in D. discoideum are nearly strand-symmetric for the background genome but show asymmetries to some degree for ncRNA genes, suggesting that a compositional contrast approach could predict the orientation of a potential ncRNA gene. However, when this possibility was explored, we did not find the predictions accurate, and we chose not to take this factor into account when we searched for novel candidates.
When we used the M1 model to search the genome sequence of D. discoideum with an expected threshold of 0.1, 2296 regions were identified that had a deviating nucleotide composition compared to the surrounding genome. The great majority of the verified and confidently predicted ncRNA genes were included in this set. The reason for the failure to identify all the C/D-box snoRNAs, and the Class I and II RNAs may be due to their relatively short length and their lack of extensive intramolecular secondary structure. Although our implementation does not explicitly impose length requirements on candidate regions, short sequences may not accumulate an aggregate score large enough to meet the significance threshold. It should be noted that the snRNA and nine tRNAs that escaped detection by the search method were masked because of an overlap with exons or repeat sequences.
It is well known that many ncRNAs occur in families with several copies of similar RNA genes spread throughout the genome. This also applies to D. discoideum ncRNA genes (Aspegren et al. 2004; Eichinger et al. 2005; Hinas et al. 2006). This observation was used to refine the search method by identifying possible families of new ncRNA genes. Of these, we confidently verified expression of four out of eight tested. To further improve the stringency of the method, we searched the flanking sequences of all the predicted ncRNA genes for a conserved sequence element, DUSE, known to be associated with D. discoideum ncRNA genes (Aspegren et al. 2004; Hinas et al. 2006; Hinas and Söderbom 2007). Two of these, notably where the sequences did not belong to a cluster but were unique in the genome, were tested for expression, and transcripts for both genes were detected. Interestingly, one of these, drd38, was found to correspond to the recently reported selenocysteine tRNA gene (Shrimali et al. 2005) that neither tRNAscan-SE nor ARAGORN identified. The observation that this unusual tRNA gene is preceded by the upstream sequence element is intriguing since this motif is not normally associated with tRNA genes. In fact, besides the selenocysteine tRNA gene, only a leucine tRNA gene seems to have the element at the expected upstream distance (data not shown). It is tempting to speculate that the presence of DUSE in front of these tRNA genes could be linked to a specific regulatory mechanism, which is distinct from the canonical tRNA genes. In addition, nine out of 12 members of one of the predicted ncRNA clusters have DUSE in close proximity, and RACE experiments identified three separate sequences within this family of which all were derived from sequences preceded by DUSE.
We have assessed the source of the performance improvement of the M1 model in D. discoideum and the potential of this extension for this purpose in other organisms, that is, Caenorhabditis elegans, Giardia lamblia, P. falciparum, and M. jannaschii, with a variety of background genomic nucleotide compositions. When we searched the AT-rich genome of M. jannaschii, previously studied by Klein et al. (2002) and Schattner (2002), we were able to detect all known as well as all predicted previously reported ncRNAs (Supplemental Data S3; Supplemental Fig. S2).
It appears that the contrast in mononucleotide content is the largest contributor to the detection of contrasting regions even when the M1 model is used. In addition, the marginal advantage of M1 in D. discoideum lies apparently in an unusual conditional dinucleotide signature in the background rather than in the targets, reflecting the fact that if we are detecting any RNA-specific dinucleotide signature, it is swamped by much more distinct signatures of the background. This was largely consistent when compared across the organisms we studied; however, we noticed an interesting consistency in certain dinucleotides, that is, AA/TT and AG/CT. These have roughly the same observed/expected dinucleotide ratio for RNAs from almost all organisms we examined but lack the corresponding consistency in background frequency ratios (Supplemental Fig. S3). Nonetheless, our investigation clearly shows that higher-order models are potentially beneficial for detecting compositionally contrasting regions of genomes in order to detect non-coding RNA genes.
In conclusion, we present a computational search for new ncRNA genes based on a screen for genomic regions with unusual dinucleotide composition. The extracted sequences are subsequently filtered with clustering of similar predicted regions and/or search for a conserved upstream sequence element. We applied this approach on the D. discoideum genome and discovered, with high confidence, new ncRNA gene candidates. Out of a total of 10 tested ncRNA candidate genes, the expression of six was verified using stringent criteria. Furthermore, when the presence of the upstream motif was used as the sole search criterion, all of the predicted genes that we tested were found to be expressed. Hence, our combined approach of using partial sum processes together with our filtering criteria gives a useful method to detect new ncRNA genes. In addition, other ways of filtering the candidates, such as clustering according to secondary structure (Will et al. 2007), could also prove important. We anticipate that our search method will be useful for the analysis of other organisms, particularly those for which genomes of closely related species have not yet been sequenced, and the use of higher-order relationships can give a marginal but valuable improvement.
Methods
Sequence data
The 2.5 release of the D. discoideum genome (Eichinger et al. 2005) along with annotations of sequence features was downloaded from the dictyBase Web site, http://dictybase.org/ (Chisholm et al. 2006) in June 2006. RepeatMasker (RepeatMasker Open-3.0) was used to mask abundant complex repeats commonly found in the intergenic sequences of D. discoideum using a custom repeat library (Glöckner et al. 2001) obtained from the Genome Sequencing Center Jena Web site at http://genome.imb-jena.de/dictyostelium/ in June 2006. In addition, a 250-kb sequence at the beginning of Chromosome 1, which is believed to function as a centromeric region (Eichinger et al. 2005), was also masked from the analyses because of its abundance of repeat sequences. Finally, exons in the dictyBase annotation were masked. Masking means that these nucleotides were changed to the letter X, leaving chromosome sequences otherwise intact. The ∼12 Mb of sequence from the six D. discoideum chromosomes remaining after the masking procedure is called the “background genome” in which contrasting regions were searched and from which, with additional masking of known or inferred ncRNA genes, background frequencies were estimated.
Search algorithm
A nucleotide sequence L of length n is transformed into a score sequence Λ according to a set of scoring rules. For the independent nucleotide case (the M0 model), each nucleotide is scored according to the set of scores {sα}, α ∈ {A, C, G, T} and similarly, for the Markov-dependent nucleotide case (the M1 model), each overlapping dinucleotide is scored based on a set of dinucleotide scores {sαβ}, αβ ∈ {AA, AC, . . . , TT}. A partial sum Sm is defined as 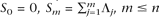 , where Λj corresponds to the score at position j in the score sequence. Then the greatest aggregate score
, where Λj corresponds to the score at position j in the score sequence. Then the greatest aggregate score
Hence, for a desired significance level, the corresponding score threshold can be determined, and, conversely, the statistical significance of a high-scoring sequence can be calculated. Specifically, it has been shown that the number of subsequences having a score greater than or equal to a score S can be approximated from a Poisson distribution according to E = nK* exp(−λ*S) (Karlin and Altschul 1990).
Both strands of the background genome data were searched at an expect value of 0.1, but for most analyses in this study, we took a region as predicted if either strand for that region was significant at the given threshold.
Scoring rules
Scores are calculated as log-odds ratios between target and background nucleotide frequencies. We derived target frequencies from inferred genome sequence parts (“genes”) corresponding to a nonredundant subset of the known ncRNA genes (Table 1). The nonredundant subset was created by removing sequences sharing >90% identity with any other sequence in the set, using the weight program from the squid v1.9g package, (S. Eddy; http://selab.janelia.org/software.html). Let Ω denote the set of different RNA families present in the training set, such that Ω = {tRNA, rRNA, snRNA, Class I RNA, Class II RNA, RNA D2, MRP RNA, RNaseP RNA, C/D-box snoRNA, H/ACA snoRNA, SRP RNA}. Let 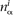 and
and 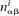 be the mononucleotide and dinucleotide counts of nucleotides α ∈ {A, C, G, T} and dinucleotides αβ ∈ {AA, AC, . . . , TT} in the ith family, i ∈ Ω, respectively; and let Mi and Di denote the total number of mononucleotides and dinucleotides in the ith family when only nucleotides and dinucleotides involving A, C, G, or T are counted. Then the mononucleotide target frequency equals
be the mononucleotide and dinucleotide counts of nucleotides α ∈ {A, C, G, T} and dinucleotides αβ ∈ {AA, AC, . . . , TT} in the ith family, i ∈ Ω, respectively; and let Mi and Di denote the total number of mononucleotides and dinucleotides in the ith family when only nucleotides and dinucleotides involving A, C, G, or T are counted. Then the mononucleotide target frequency equals 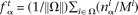 and the dinucleotide target frequency equals
and the dinucleotide target frequency equals 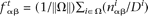 .
.
In calculations of the background frequencies, we used the previously defined background genome in which all known or confidently predicted ncRNA genes have additionally been masked. Using the notation above, let nα and nαβ be the count of nucleotide α and dinucleotide αβ, respectively, and let M and D be the total number of mononucleotides and dinucleotides, respectively. The background mononucleotide and dinucleotide background frequencies were calculated according to 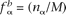 and
and 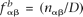 , respectively. The score associated with the nucleotide α was then calculated as the log-likelihood ratio
, respectively. The score associated with the nucleotide α was then calculated as the log-likelihood ratio 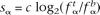 , where c is a scaling constant introduced for practical purposes (we used a value of c = 10) and the scores were rounded to the closest integer. Similarly, the score for the dinucleotide αβ was calculated as sαβ = c log2(qαβ/pαβ), where pαβ and qαβ are the probabilities of making a transition from state α to state β in the Markov chain and can be calculated according to
, where c is a scaling constant introduced for practical purposes (we used a value of c = 10) and the scores were rounded to the closest integer. Similarly, the score for the dinucleotide αβ was calculated as sαβ = c log2(qαβ/pαβ), where pαβ and qαβ are the probabilities of making a transition from state α to state β in the Markov chain and can be calculated according to 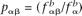 for the background and
for the background and 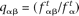 for the target. In addition, a set of custom scores was introduced to accommodate masked segments according to sγ = sαγ = sγβ = sγδ = −∞, α, β ≠ X, γ, δ = X (the letter X indicates a masked nucleotide). Ambiguous or unrecognized nucleotides encountered were scored according to sγ = minαsα, sαγ = minβsαβ, sγβ = minαsαβ, sγδ = minαβsαβ, α, β ∈ {A, C, G, T}, γ, δ ∈ {R, Y, M, K, S, W, H, B, V, D, N} (the set of IUPAC ambiguous nucleotide symbols).
for the target. In addition, a set of custom scores was introduced to accommodate masked segments according to sγ = sαγ = sγβ = sγδ = −∞, α, β ≠ X, γ, δ = X (the letter X indicates a masked nucleotide). Ambiguous or unrecognized nucleotides encountered were scored according to sγ = minαsα, sαγ = minβsαβ, sγβ = minαsαβ, sγδ = minαβsαβ, α, β ∈ {A, C, G, T}, γ, δ ∈ {R, Y, M, K, S, W, H, B, V, D, N} (the set of IUPAC ambiguous nucleotide symbols).
Model validation and selection
ROC plots were calculated by running the M0 and M1 models on simulated data sets for which known or confidently predicted ncRNA genes were inserted at their natural positions into background genome data generated according to a fifth-order Markov model of the background genome data (estimated from the same data as the background model for the score matrices). The same pattern of feature masking as the natural data was retained in the simulated data. A true positive was counted if one or more significant regions overlapped it with at least one nucleotide. A true negative was counted if no significant region overlapped it. If a significant region overlapped a true negative and simultaneously overlapped a true positive on one side, the true negative was still counted as such. Sensitivity was calculated as Sn = [TP/(TP + FN)], where TP and FN denote the number of true positives and false negatives, respectively. Specificity was calculated as Sp = [TN/(TN + FP)], where TN and FP denote the number of true negatives and false positives, respectively. Since we only had a limited amount of nonredundant target sequences, we chose to do a cross-validation analysis with two folds to have true positive examples representing diverse classes of ncRNAs in each fold. We divided the nonredundant target set of known ncRNAs in two halves, and the background set was divided in the same way. The first half of the ncRNA set was used together with one-half of the background set for calculating the scores, and the other half was randomly inserted into the other half of the background set for validation. This procedure was repeated for the two halves. We used both the artificial background described above and real data consisting of the nonrepetitive, intergenic regions from D. discoideum, assuming that no unknown true ncRNA genes overlapped those. ROC-plot analyses were performed, and the mean and standard deviation of the AUC was calculated.
Upstream sequence elements
In order to determine if a DUSE was associated with a candidate gene, we searched for the presence of sequence elements AC CCATAA or TCCCAHAA (H = A, C, or T) within the 150 nt flanking the candidate.
Sequence clustering
Sequence clusters were built by single-linkage clustering using the BLASTCLUST v2.2.9 software. Neighbors in a cluster were required to share at least 80% sequence identity covering a minimum of 50% of one of the sequence’s length. The word size was set to 11, and the dust low-complexity filter was used.
Bioinformatics tools
Version 1.1 of the tRNA gene prediction software ARAGORN was used for prediction of tRNA genes (Laslett and Canbäck 2004). The RepeatMasker software, version Open-3.1.2, was used for masking of interspersed repeats. Clustering of candidates was performed using the BLASTCLUST v2.2.9 software, available in the BLAST package at http://www.ncbi.nlm.nih.gov/BLAST/download.shtml. Secondary-structure predictions were performed using RNAfold and RNAalifold in version 1.6.2 of the Vienna RNA Package, available at http://www.tbi.univie.ac.at/~ivo/RNA/ (Hofacker et al. 1994, 2002). The predicted secondary structures were manually edited using the RNAviz v2.0 editor, available at http://rnaviz.sourceforge.net/ (De Rijk et al. 2003). Multiple sequence alignments were constructed using ClustalW v1.83 (Thompson et al. 1994).
Java routines and source code
We have bundled the Java routines used for calculation of score matrices and statistical parameters as well as identification of high-scoring regions of contrasting composition into a package named SIGRS (Single Genome ncRna Search). For class files and source code, see Supplemental Data S4.
Sequence data
Experimentally verified ncRNAs have been submitted to dictyBase (http://dictybase.org). An annotation file compatible with the dictyBase genome browser that can be used to visualize all predictions in their genomic context has been compiled (Supplemental Data S5), and a fasta file with nucleotide sequences of the predictions is available (Supplemental Data S6).
Oligonucleotides
Sequences of the DNA oligonucleotides (Invitrogen) used in this study can be found in Supplemental Table S2. Sequences of the RNA oligonucleotides used for 5′- and 3′-RACE (Dharmacon) were described in Aspegren et al. (2004).
Growth conditions
D. discoideum strain AX4 (Knecht et al. 1986) was grown axenically in HL5 medium and developed on nitrocellulose membranes (Sussman 1987).
Expression analysis
Total RNA was extracted from axenically growing cells and cells developed for 16 and 24 h, respectively, by the TRIzol method (Invitrogen). For Northern blot analysis, 20 μg RNA was separated on an 8% polyacrylamide/7 M urea/1× TBE gel and electroblotted to a Hybond N+ membrane (GE Healthcare) together with radiolabeled markers pUC19/MspI (Fermentas) and RNA decade marker (Ambion). The membranes were hybridized overnight with 32P-end-labeled DNA oligonucleotides at 42°C in Church buffer (7% SDS, 0.5 M NaPO4 pH 7.2, 1 mM EDTA, and 1% BSA) and subsequently washed twice for 5 min in 2× SSC/0.1% SDS, twice for 10 min in 1× SSC/0.1% SDS, and twice for 5 min in 0.5× SSC/0.1% SDS at 42°C, followed by washing twice for 5 min in 0.5× SSC/0.1%SDS at 50°C. Hybridization signals were detected by a PhosphorImager (Molecular Dynamics). For determination of RNA termini, 5′ and 3′ RACE were carried out as described previously (Aspegren et al. 2004). Candidate RNAs were regarded to be recognized by the oligonucleotide probes only if they were fully complementary.
Deposited sequences
Primary sequences for ncRNAs drf9_3 and drf9_7 determined by 3′-RACE analysis have been deposited in the GenBank database. The accession numbers are GenBank: EF551319 for drf9_3 and GenBank: EF551320 for drf9_7.
Acknowledgments
We thank Lotta Avesson and Björn Garpefjord for their contributions on the experimental validation of candidate ncRNAs and Johan Reimegård for valuable suggestions. dictyBase (http://dictybase.org/) is also gratefully acknowledged. This work was supported by grants from the European Community (FOSRAK, EC005120) to F.S. and from Wallenberg Consortium North Foundation, The Swedish Research Council to Uppsala RNA Research Center in the form of a Linnéstöd and Swedish Foundation for Strategic Research to L.A.K.
References
- ↵S.F. Altschul,W. Gish,W. Miller,E.W. Myers,D.J. Lipman,(1990) Basic local alignment search tool. J. Mol. Biol. 215:403–410.
- ↵S.F. Altschul,T.L. Madden,A.A. Schaffer,J. Zhang,Z. Zhang,W. Miller,D.J. Lipman,(1997) Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402.
- ↵A. Aravin,T. Tuschl,(2005) Identification and characterization of small RNAs involved in RNA silencing. FEBS Lett. 579:5830–5840.
- ↵A. Aspegren,A. Hinas,P. Larsson,A. Larsson,F. Söderbom,(2004) Novel non-coding RNAs in Dictyostelium discoideum and their expression during development. Nucleic Acids Res. 32:4646–4656.
- ↵S.L. Baldauf,A.J. Roger,I. Wenk-Siefert,W.F. Doolittle,(2000) A kingdom-level phylogeny of eukaryotes based on combined protein data. Science 290:972–977.
- ↵D.S. Chew,M.Y. Leung,K.P. Choi,(2007) AT excursion: A new approach to predict replication origins in viral genomes by locating AT-rich regions. BMC Bioinformatics 8:163, 10.1186/1471-2105-8-163.
- ↵R.L. Chisholm,P. Gaudet,E.M. Just,K.E. Pilcher,P. Fey,S.N. Merchant,W.A. Kibbe,(2006) dictyBase, the model organism database for Dictyostelium discoideum. Nucleic Acids Res. 34:D423–D427, 10.1093/nar/gkj090.
- ↵P. Clote,F. Ferre,E. Kranakis,D. Krizanc,(2005) Structural RNA has lower folding energy than random RNA of the same dinucleotide frequency. RNA 11:578–591.
- ↵M. Csűrös,(2004) Maximum-scoring segment sets. IEEE/ACM Trans. Comput. Biol. Bioinform. 1:139–150.
- ↵P. De Rijk,J. Wuyts,R. De Wachter,(2003) RnaViz 2: An improved representation of RNA secondary structure. Bioinformatics 19:299–300.
- ↵S.R. Eddy,(2002) Computational genomics of noncoding RNA genes. Cell 109:137–140.
- ↵S. Edvardsson,P.P. Gardner,A.M. Poole,M.D. Hendy,D. Penny,V. Moulton,(2003) A search for H/ACA snoRNAs in yeast using MFE secondary structure prediction. Bioinformatics 19:865–873.
- ↵L. Eichinger,J.A. Pachebat,G. Glockner,M.A. Rajandream,R. Sucgang,M. Berriman,J. Song,R. Olsen,K. Szafranski,Q. Xu,(2005) The genome of the social amoeba Dictyostelium discoideum. Nature 435:43–57.
- ↵M.J. Gardner,N. Hall,E. Fung,O. White,M. Berriman,R.W. Hyman,J.M. Carlton,A. Pain,K.E. Nelson,S. Bowman,(2002) Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419:498–511.
- ↵G. Glöckner,K. Szafranski,T. Winckler,T. Dingermann,M.A. Quail,E. Cox,L. Eichinger,A.A. Noegel,A. Rosenthal,(2001) The complex repeats of Dictyostelium discoideum. Genome Res. 11:585–594.
- ↵S. Griffiths-Jones,S. Moxon,M. Marshall,A. Khanna,S.R. Eddy,A. Bateman,(2005) Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33:D121–D124, 10.1093/nar/gki081.
- ↵J.H. Havgaard,R.B. Lyngso,G.D. Stormo,J. Gorodkin,(2005) Pairwise local structural alignment of RNA sequences with sequence similarity less than 40%. Bioinformatics 21:1815–1824.
- ↵A. Hinas,F. Söderbom,(2007) Treasure hunt in an amoeba: Non-coding RNAs in Dictyostelium discoideum. Curr. Genet. 51:141–159.
- ↵A. Hinas,P. Larsson,L. Avesson,L.A. Kirsebom,A. Virtanen,F. Söderbom,(2006) Identification of the major spliceosomal RNAs in Dictyostelium discoideum reveals developmentally regulated U2 variants and polyadenylated snRNAs. Eukaryot. Cell 5:924–934.
- ↵I.L. Hofacker,W. Fontana,P.F. Stadler,L.S. Bonhoeffer,M. Tacker,P. Schuster,(1994) Fast folding and comparison of RNA secondary structures. Monatsh. Chem. 125:167–188.
- ↵I.L. Hofacker,M. Fekete,P.F. Stadler,(2002) Secondary structure prediction for aligned RNA sequences. J. Mol. Biol. 319:1059–1066.
- ↵A. Hüttenhofer,J. Vogel,(2006) Experimental approaches to identify non-coding RNAs. Nucleic Acids Res. 34:635–646.
- ↵S. Karlin,S.F. Altschul,(1990) Methods for assessing the statistical significance of molecular sequence features by using general scoring schemes. Proc. Natl. Acad. Sci. 87:2264–2268.
- ↵S. Karlin,V. Brendel,(1992) Chance and statistical significance in protein and DNA sequence analysis. Science 257:39–49.
- ↵S. Karlin,C. Burge,(1995) Dinucleotide relative abundance extremes: A genomic signature. Trends Genet. 11:283–290.
- S. Karlin,A. Dembo,(1992) Limit distributions of maximal segmental score among Markov-dependent partial sums. Adv. Appl. Probab. 24:113.
- S. Karlin,A. Dembo,T. Kawabata,(1990) Statistical composition of high-scoring segments from molecular sequences. Ann. Statist. 18:571–581.
- ↵S. Karlin,A.M. Campbell,J. Mrazek,(1998) Comparative DNA analysis across diverse genomes. Annu. Rev. Genet. 32:185–225.
- ↵R.J. Klein,Z. Misulovin,S.R. Eddy,(2002) Noncoding RNA genes identified in AT-rich hyperthermophiles. Proc. Natl. Acad. Sci. 99:7542–7547.
- ↵D.A. Knecht,S.M. Cohen,W.F. Loomis,H.F. Lodish,(1986) Developmental regulation of Dictyostelium discoideum actin gene fusions carried on low-copy and high-copy transformation vectors. Mol. Cell. Biol. 6:3973–3983.
- ↵M. Kuhlmann,B.E. Borisova,M. Kaller,P. Larsson,D. Stach,J. Na,L. Eichinger,F. Lyko,V. Ambros,F. Söderbom,(2005) Silencing of retrotransposons in Dictyostelium by DNA methylation and RNAi. Nucleic Acids Res. 33:6405–6417.
- ↵C. Kyriakopoulou,P. Larsson,L. Liu,J. Schuster,F. Söderbom,L.A. Kirsebom,A. Virtanen,(2006) U1-like snRNAs lacking complementarity to canonical 5′ splice sites. RNA 12:1603–1611.
- ↵D. Laslett,B. Canbäck,(2004) ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32:11–16.
- ↵T.M. Lowe,S.R. Eddy,(1997) tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964.
- ↵T.M. Lowe,S.R. Eddy,(1999) A computational screen for methylation guide snoRNAs in yeast. Science 283:1168–1171.
- S.M. Marquez,J.K. Harris,S.T. Kelley,J.W. Brown,S.C. Dawson,E.C. Roberts,N.R. Pace,(2005) Structural implications of novel diversity in eucaryal RNase P RNA. RNA 11:739–751.
- ↵D.H. Mathews,(2006) Revolutions in RNA secondary structure prediction. J. Mol. Biol. 359:526–532.
- ↵I.M. Meyer,(2007) A practical guide to the art of RNA gene prediction. Brief Bioinform. 8:396–414.
- ↵J.S. Pedersen,G. Bejerano,A. Siepel,K. Rosenbloom,K. Lindblad-Toh,E.S. Lander,J. Kent,W. Miller,D. Haussler,(2006) Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput. Biol. 2:e33, 10.1371/journal.pcbi.0020033.
- ↵P. Piccinelli,M.A. Rosenblad,T. Samuelsson,(2005) Identification and analysis of ribonuclease P and MRP RNA in a broad range of eukaryotes. Nucleic Acids Res. 33:4485–4495.
- ↵E. Rivas,S.R. Eddy,(2000) Secondary structure alone is generally not statistically significant for the detection of noncoding RNAs. Bioinformatics 16:583–605.
- ↵E. Rivas,S. Eddy,(2001) Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatics 2:8, 10.1186/1471-2105-2-8.
- ↵P. Schattner,(2002) Searching for RNA genes using base-composition statistics. Nucleic Acids Res. 30:2076–2082.
- ↵P. Schattner,S. Barberan-Soler,T.M. Lowe,(2006) A computational screen for mammalian pseudouridylation guide H/ACA RNAs. RNA 12:15–25.
- ↵R.K. Shrimali,A.V. Lobanov,X.-M. Xu,M. Rao,B.A. Carlson,D.C. Mahadeo,C.A. Parent,V.N. Gladyshev,D.L. Hatfield,(2005) Selenocysteine tRNA identification in the model organisms Dictyostelium discoideum and Tetrahymena thermophila. Biochem. Biophys. Res. Commun. 329:147–151.
- R. Sucgang,G. Chen,W. Liu,R. Lindsay,J. Lu,D. Muzny,G. Shaulsky,W. Loomis,R. Gibbs,A. Kuspa,(2003) Sequence and structure of the extrachromosomal palindrome encoding the ribosomal RNA genes in Dictyostelium. Nucleic Acids Res. 31:2361–2368.
- ↵M. Sussman,(1987) Cultivation and synchronous morphogenesis of Dictyostelium under controlled experimental conditions. Methods Cell Biol. 28:9–29.
- ↵J.D. Thompson,D.G. Higgins,T.J. Gibson,(1994) CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680.
- ↵D.H. Turner,N. Sugimoto,(1988) RNA structure prediction. Annu. Rev. Biophys. Biophys. Chem. 17:167–192.
- ↵R. Upadhyay,P. Bawankar,D. Malhotra,S. Patankar,(2005) A screen for conserved sequences with biased base composition identifies noncoding RNAs in the A-T rich genome of Plasmodium falciparum. Mol. Biochem. Parasitol. 144:149–158.
- ↵S. Washietl,I.L. Hofacker,P.F. Stadler,(2005) Fast and reliable prediction of noncoding RNAs. Proc. Natl. Acad. Sci. 102:2454–2459.
- ↵C.L. Will,R. Lührmann,(2001) Spliceosomal UsnRNP biogenesis, structure and function. Curr. Opin. Cell Biol. 13:290–301.
- ↵S. Will,K. Reiche,I.L. Hofacker,P.F. Stadler,R. Backofen,(2007) Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 3:e65, 10.1371/journal.pcbi.0030065.
- J.A. Wise,A.M. Weiner,(1981) The small nuclear RNAs of the cellular slime mold Dictyostelium discoideum. Isolation and characterization. J. Biol. Chem. 256:956–963.
- ↵C. Workman,A. Krogh,(1999) No evidence that mRNAs have lower folding free energies than random sequences with the same dinucleotide distribution. Nucleic Acids Res. 27:4816–4822.
- ↵T. Xia,J. SantaLucia,M.E. Burkard,R. Kierzek,S.J. Schroeder,X. Jiao,C. Cox,D.H. Turner,(1998) Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry 37:14719–14735.
- ↵J.H. Zar,(1999) Biostatistical analysis (Prentice-Hall, Upper Saddle River, NJ).
Notes
[1] [Supplemental material is available online at http://www.genome.org. The sequence data from this study have been submitted to GenBank under accession nos. EF551319 and EF551320.]
[2] Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.069104.107.