
ALLPATHS assemblies of simulated 30-base microreads
Click on table to view larger version.
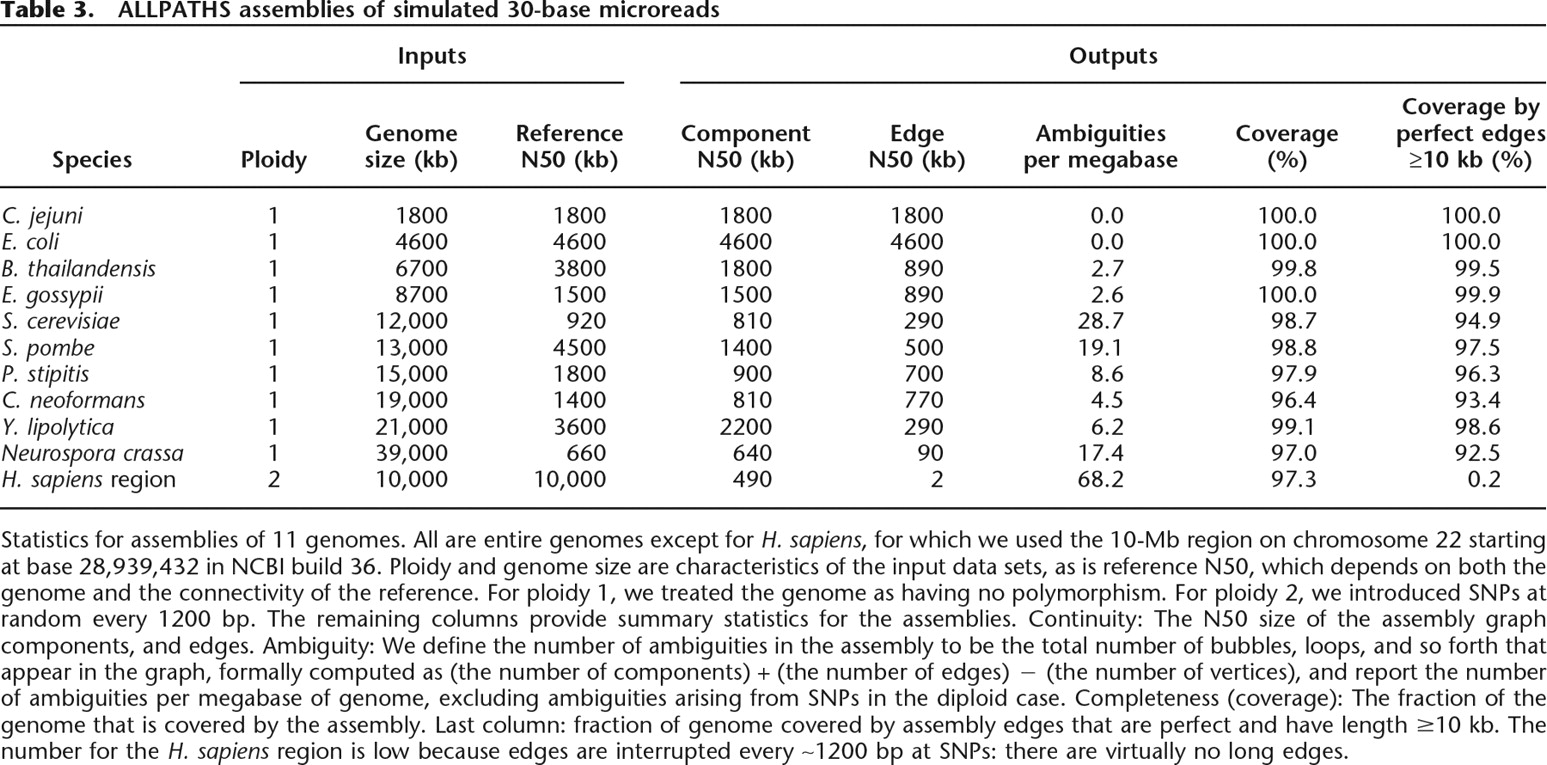
Statistics for assemblies of 11 genomes. All are entire genomes except for H. sapiens, for which we used the 10-Mb region on chromosome 22 starting at base 28,939,432 in NCBI build 36. Ploidy and genome size are characteristics of the input data sets, as is reference N50, which depends on both the genome and the connectivity of the reference. For ploidy 1, we treated the genome as having no polymorphism. For ploidy 2, we introduced SNPs at random every 1200 bp. The remaining columns provide summary statistics for the assemblies. Continuity: The N50 size of the assembly graph components, and edges. Ambiguity: We define the number of ambiguities in the assembly to be the total number of bubbles, loops, and so forth that appear in the graph, formally computed as (the number of components) + (the number of edges) − (the number of vertices), and report the number of ambiguities per megabase of genome, excluding ambiguities arising from SNPs in the diploid case. Completeness (coverage): The fraction of the genome that is covered by the assembly. Last column: fraction of genome covered by assembly edges that are perfect and have length ≥10 kb. The number for the H. sapiens region is low because edges are interrupted every ∼1200 bp at SNPs: there are virtually no long edges.








