
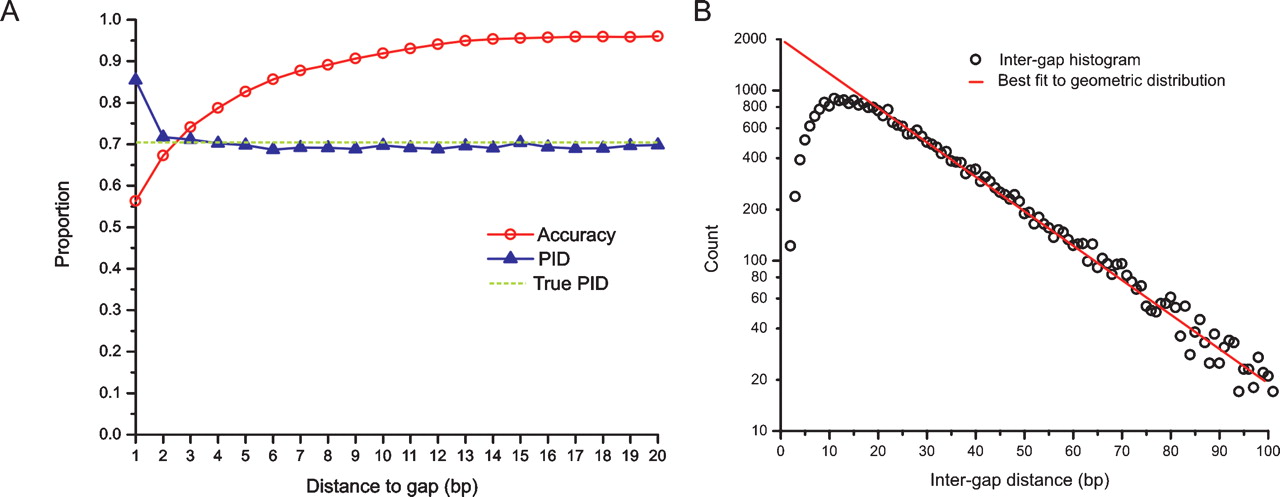
Effects of alignment biases in relation to gaps. Alignment biases cause systematic errors in alignments that are non-uniformly distributed with respect to alignment gaps. (A, left) The proportion sequence identity (PID, blue triangles), the true PID (dashed), and the proportion of correctly aligned columns (accuracy, red circles), for realigned sequences evolving under a Jukes–Cantor model, as a function of the distance to the nearest gap in the inferred alignment. The spuriously high PID and low accuracy adjacent to gaps is caused by gap wander. Gap annihilation is responsible for the reduced accuracy, and the slight reduction of PID below the true value away from gaps. (B, right) A histogram of intergap distances (circles), and the best fit to a geometric distribution (red line). The scarcity of closely spaced gaps (less than about 20 nucleotides apart) is due to gap attraction and affects a large number of gaps (note the logarithmic scale).








