
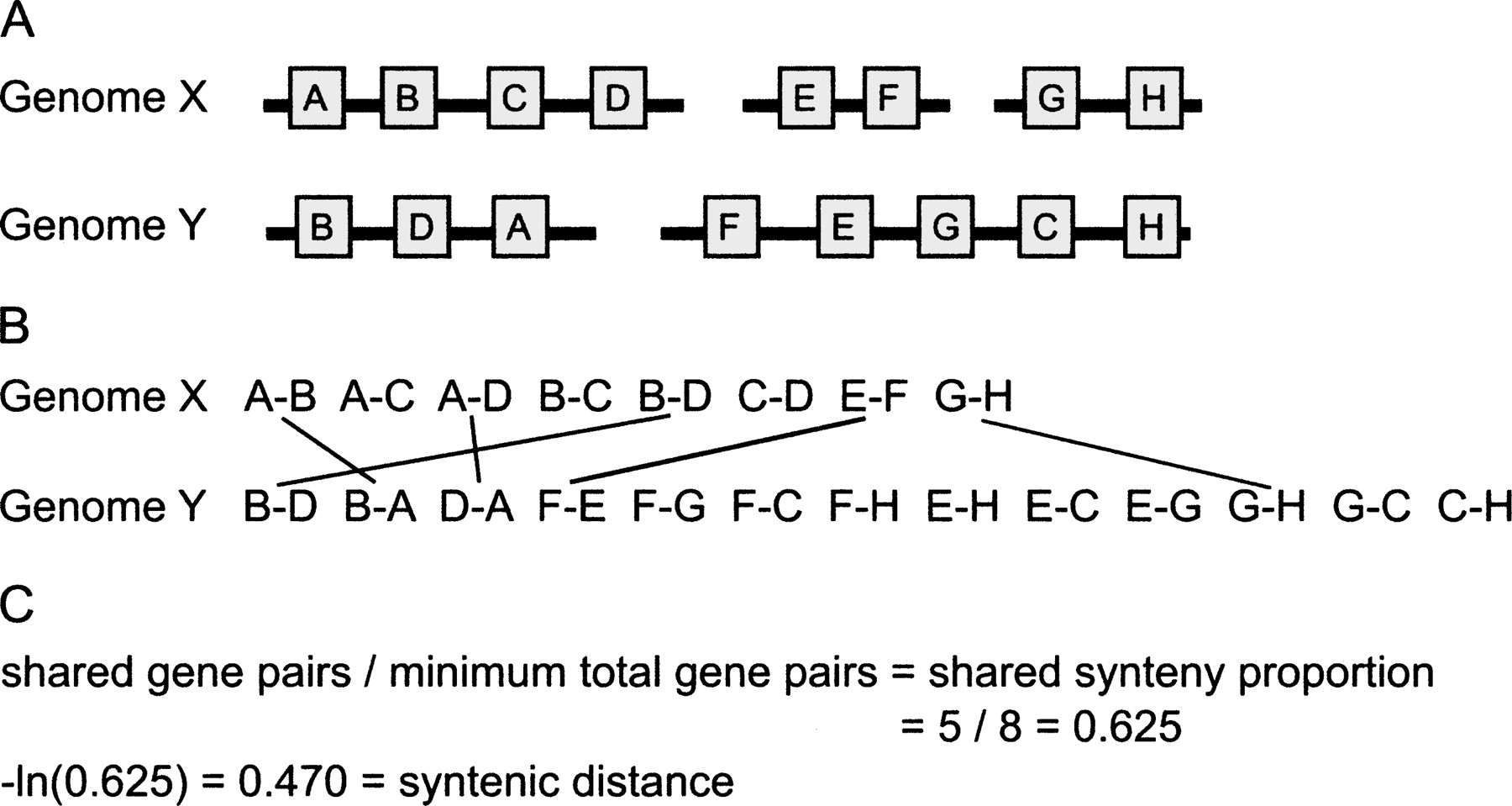
Estimating the amount of synteny conservation between two genomes. This figure illustrates the method we use to calculate the “syntenic distance” between pairs of genomes. (A) Two genomes, X and Y, share eight orthologous genes, present on three genome fragments in the genome X, and two in genome Y. (B) These orthologous genes can be decomposed into proximate gene pairs (see Methods and Fig. 1). (C) From these gene pairs, we can calculate the shared synteny proportion and convert this proportion into a time-linear distance measure by taking the negative natural logarithm. This is a highly simplified case—for analyses of real genomes, genome fragments are required to have at least 10 genes.








