
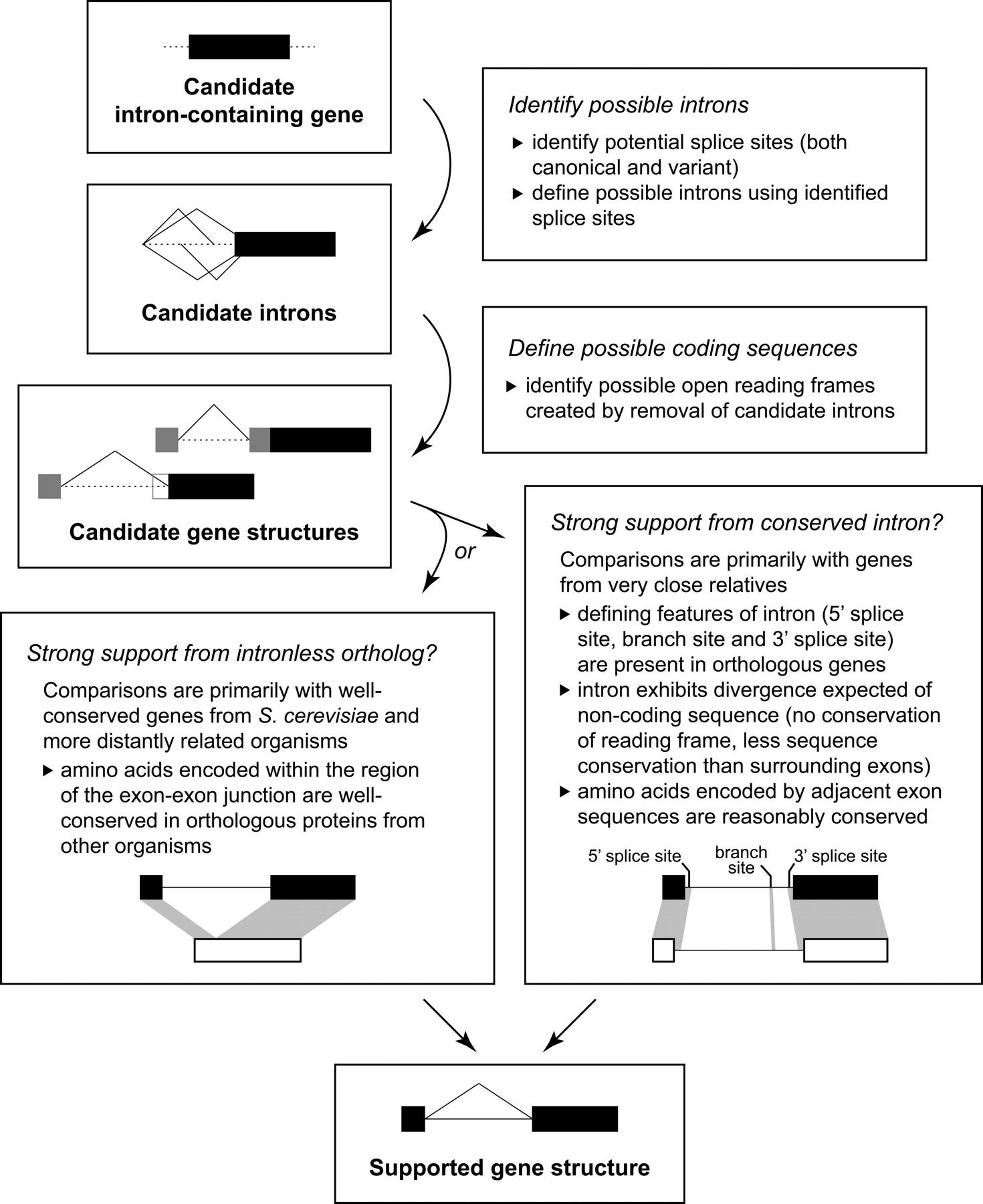
Refinement and confirmation of candidate introns. Once candidate intron-containing genes are identified by any of our methods, we use a manual bioinformatics approach to identify possible introns and determine whether any are strongly supported by available phylogenetic data. Because our approach does not require annotated sequences for comparison, we can rely on support from the sequenced genomes of other Candida species when protein conservation is insufficient for comparisons with more distant relatives. One of our two sets of criteria—strong support from either orthologous coding sequences or conserved introns—must be met for us to consider an intron supported. (See Methods for further details.)








