
Table 2.
Results for B. bacteriovorus simulation
Click on table to view larger version.
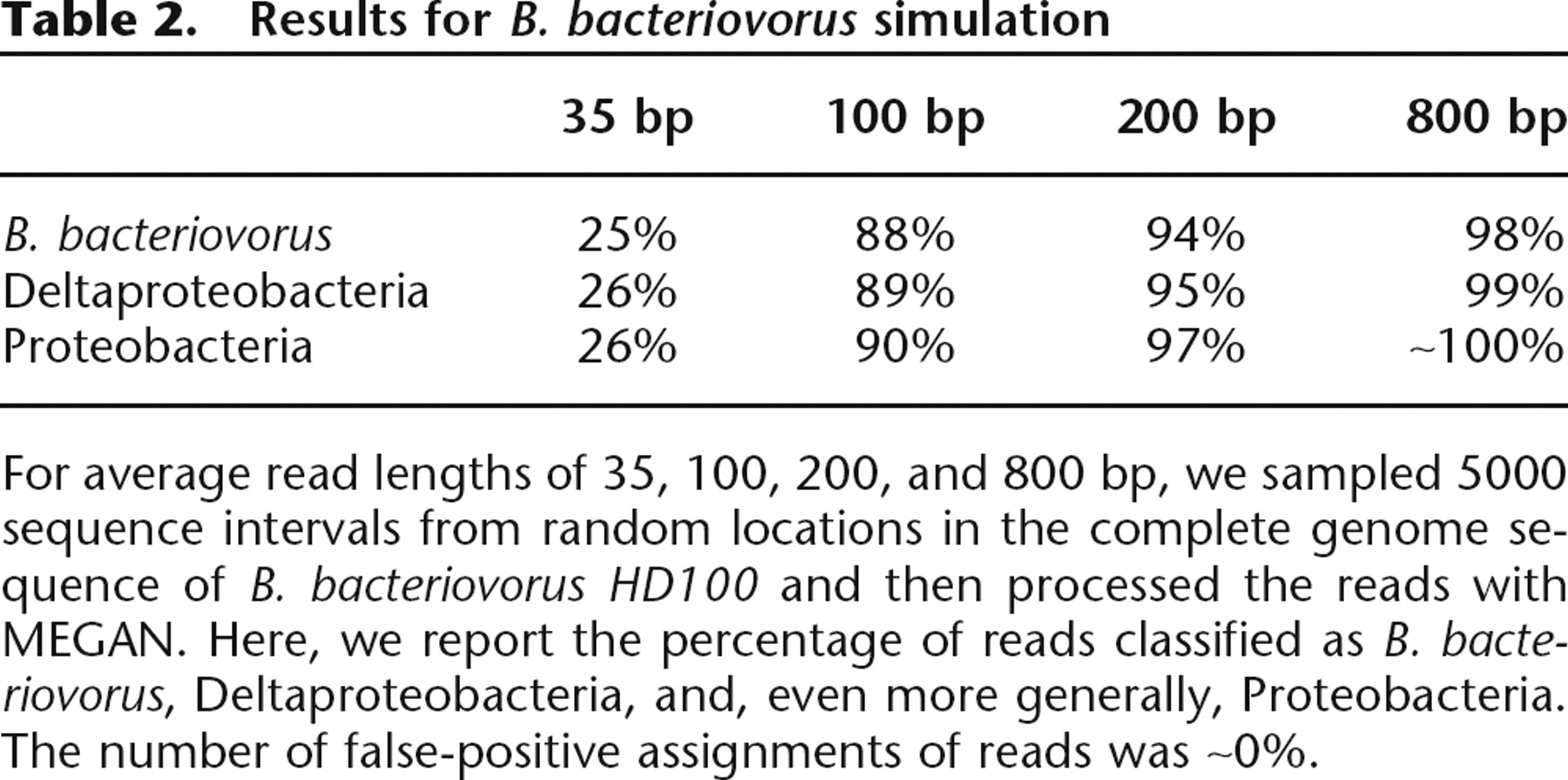
For average read lengths of 35, 100, 200, and 800 bp, we sampled 5000 sequence intervals from random locations in the complete genome sequence of B. bacteriovorus HD100 and then processed the reads with MEGAN. Here, we report the percentage of reads classified as B. bacteriovorus, Deltaproteobacteria, and, even more generally, Proteobacteria. The number of false-positive assignments of reads was ∼0%.








