
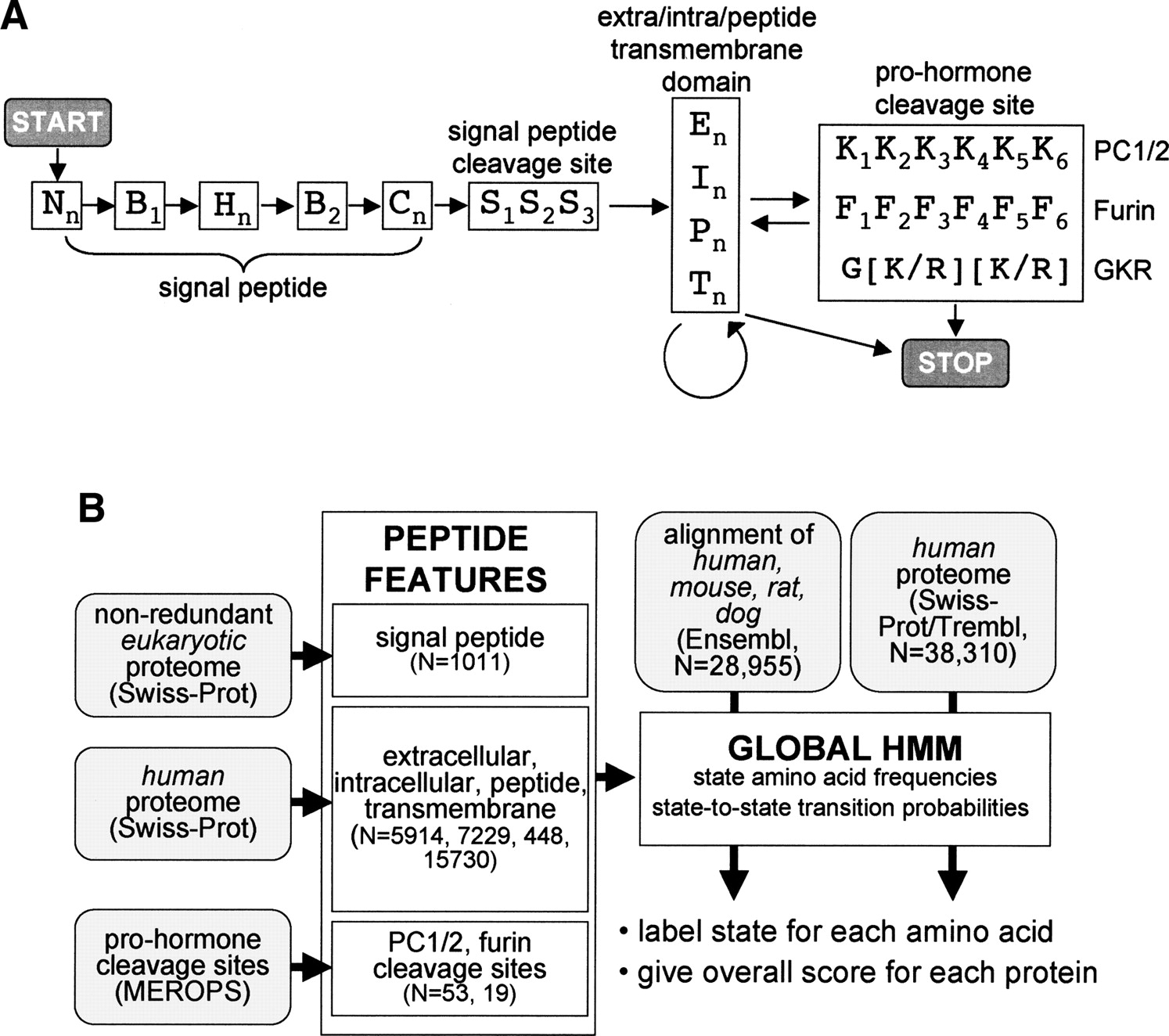
Hidden Markov model (HMM) for the identification of peptide hormones. (A) State structure of the peptide hormone HMM with states indicated by letters and transitions between states indicated by arrows. States with numerical subscripts are single amino acid states, while states with the “n” subscript are multiple amino acid states whose length is determined by the transition probability between that state and other permitted states. Nn, B1, Hn, B2, Cn, and S1–3, are N terminus, border, hydrophobic, C terminus, and cleavage site states, respectively, of the signal peptide feature. En, In, Pn, and Tn are extracellular, intracellular, peptide, and transmembrane states, respectively, while K1–6 and F1–6 are pro-hormone cleavage site states and G[K/R][K/R] is a simple sequence motif. START and STOP mark entry and exit points of the HMM. (B) Protocol for building and running the peptide hormone HMM. HMM states for individual sequence features were built by learning amino acid frequencies and transition probabilities from sets of proteins or motifs with known features (N = size of training set). Signal peptide states were built using a previously curated set of eukaryotic SWISS-PROT proteins; extra/intra/peptide/transmembrane states were built using selected sets of human SWISS-PROT proteins; and pro-hormone cleavage sites were built using a set of PC1/2 and furin sites from the MEROPS database. The peptide hormone HMM was assembled with the state-to-state transition constraints outlined in A. Finally, the HMM was used to assign states and scores to either a set of alignments of human, mouse, rat, and dog proteins from Ensembl or a set of human proteins from SWISS-PROT/TrEMBL.








