
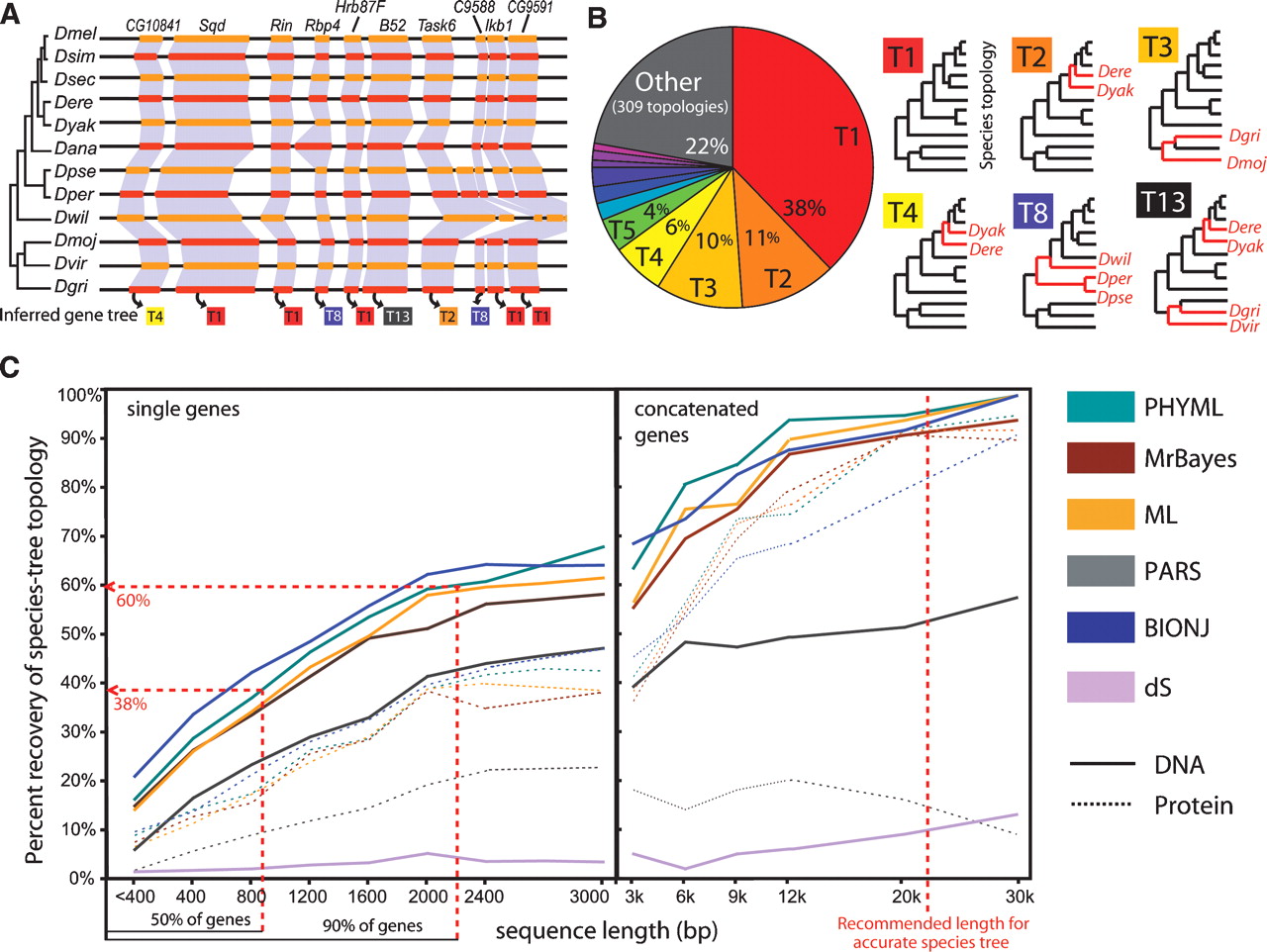
Large-scale gene-tree incongruence correlates with gene length. (A) Unambiguously orthologous genes in syntenic regions show diverse PHYML trees, even for consecutive genes (topologies numbered according to their genome-wide frequency). (B) Frequency and topology of most abundant ML gene trees for syntenic orthologs across 12 fly genomes. Discrepancies from the species topology (red branches) correspond to rotations of short internal branches. (C) Percentage of gene trees congruent to the species phylogeny correlates with gene length, regardless of the method used. DNA-based reconstruction, which uses threefold as many aligned positions, consistently outperformed protein-based reconstruction across all methods.








